
Abstract
Incorporation of nanoparticles composed of surface-functionalized fumed silica (FS) or native colloidal silica (CS) into a nanostructured block copolymer yields hybrid nanocomposites whose mechanical properties can be tuned by nanoparticle concentration and surface chemistry. In this work, dynamic rheology is used to probe the frequency and thermal responses of nanocomposites composed of a symmetric poly(styrene-b-methyl methacrylate) (SM) diblock copolymer and varying in nanoparticle concentration and surface functionality. At sufficiently high loading levels, FS nanoparticle aggregates establish a load-bearing colloidal network within the copolymer matrix. Transmission electron microscopy images reveal the morphological characteristics of the nanocomposites under these conditions.
Similar content being viewed by others

Introduction
Block copolymers remain one of the most extensively studied classes of polymers due to their innate ability to infuse the dissimilar properties of homopolymers into a single material by spontaneously self-organizing at the molecular level. Molecular self-assembly is a direct consequence of thermodynamic incompatibility between the contiguous sequences comprising a block copolymer and results in the formation of (a)periodic nanoscale morphologies that can be tailored for diverse (nano)technologies [1, 2]. Ordered morphologies observed in simple AB diblock copolymers include A(B) spheres on a body- or face-centered cubic lattice in a B(A) matrix, A(B) cylinders on a hexagonal lattice in a B(A) matrix, triply periodic bicontinuous channels or alternating lamellar sheets. Targeted addition of a selective low-molar-mass solvent or relatively low-molecular-weight homopolymer to an ordered block copolymer can be used to preferentially swell one of the domains comprising the copolymer nanostructure and ultimately yield tunable transitions to morphologies with specific mechanical or spatial properties [3–8]. Recent studies have extended this general design paradigm by modifying ordered block copolymers with surface-functionalized inorganic nanoparticles to achieve hybrid nanocomposites. Unlike conventional nanocomposites prepared from homopolymers, block copolymer nanocomposites rely on the existing copolymer nanostructure to template—that is, spatially modulate—the nanoparticles.
Previous experimental studies [8–12] of block copolymer nanocomposites have focused on the precise positioning of nanoparticles within the copolymer nanostructure for use in optics, such as waveguides. Such efforts have demonstrated that, if sufficiently small with respect to the host copolymer molecules (i.e., the characteristic size of the copolymer nanostructure), nonselective nanoparticles tend to distribute uniformly throughout the copolymer matrix in much the same fashion as nonselective solvent molecules. Selective nanoparticles, on the other hand, tend to locate along the interface separating adjacent domains within the copolymer nanostructure due to interfacial energy considerations, which result in fewer contacts between A and B repeat units. Larger selective nanoparticles are enthalpically driven to the core of the compatible domains to minimize repulsive contacts with incompatible blocks. Surface-functionalized nanoparticles have likewise been incorporated into ordered block copolymers to promote changes in morphology [13–15], as well as changes in phase behavior [16–18] discerned from the order–disorder transition (ODT). We have recently demonstrated [18] that the ODT of a poly(styrene-b-methyl methacrylate) (SM) diblock copolymer modified with surface-functionalized fumed silica (FS) or native (hydroxyl-terminated) colloidal silica (CS) decreases monotonically with increasing nanoparticle loading. If, however, oligostyrene-functionalized CS is added to the copolymer, the ODT increases slightly before dropping, in qualitative agreement with self-consistent field predictions.
The objective of the present work is to examine how the mechanical properties of such block copolymer nanocomposites evolve as the concentration of surface-functionalized FS and native CS is systematically increased. Dynamic melt rheology is employed to investigate the mechanical properties, and transmission electron microscopy (TEM) is used to examine the morphology of one of the nanocomposites.
Experimental
The SM copolymer was synthesized via sequential living anionic polymerization of the S block in cyclohexane at 60°C, followed by the M block in tetrahydrofuran at −78°C, with sec-butyllithium as the initiator. According to proton nuclear magnetic resonance (1H NMR) spectroscopy and size-exclusion chromatography (SEC), the block masses measured 13,000 each, with an overall polydispersity of 1.05. Three grades of functionalized FS were obtained in powder form from Degussa Corp. (Parsippany, NJ) and probed the effects of hydrophilicity versus hydrophobicity and block selectivity: hydroxyl-terminated (OH), methacrylate-terminated (MA) and octyl-terminated (C8). According to the manufacturer, the primary particle size in each case was ~12 nm. The CS nanoparticles with an average diameter of 10–15 nm were provided as a suspension (20% solids) in dimethylacetamide by Nissan Chemicals (Houston, TX). Specimens for dynamic melt rheology were produced by ultrasonicating the nanoparticles (at a specimen-specific concentration relative to the copolymer) for 30 min in toluene to achieve a satisfactory dispersion, followed by copolymer dissolution and further ultrasonication, and then air- and vacuum-drying, all performed at ambient temperature. No copolymer degradation due to ultrasonication was detected according to SEC analysis of the resultant films.
Dynamic rheology was performed on an ARES strain-controlled rheometer equipped with serrated 8 mm parallel plates and operated at 2% strain amplitude to ensure linear viscoelasticity. Disks measuring 8 mm in diameter and 1 mm thick were melt-pressed at 150°C and heated to 220°C under nitrogen. Frequency (ω) spectra were acquired at discrete temperatures above and below the ODT, while isochronal temperature sweeps were performed at ω = 1 rad/s and a cooling rate of 1°C/min under a nitrogen purge to avoid oxidative degradation. Specimens for TEMT were prepared by sectioning the glassy nanocomposites at ambient temperature. Electron-transparent sections measuring ca. 150 nm thick were subjected to the vapor of 0.5% RuO4(aq) to selectively stain the styrenic units. Serial TEM tilt images were collected on a Gatan UltraScan 4000 CCD camera at a resolution of 0.76 nm/pixel and tilt angles ranging from −69° to +69° at an angular interval of 1.5° on a Technai T20 microscope operated at 200 kV. While the full tilt series was aligned using a pre-calibrated geometric model based on a high-precision goniometer stage [19], only representative images acquired at 0°, 15° and 30° are reported herein.
Results and Discussion
According to the discontinuous change in the dynamic storage modulus (G′) encountered during an isochronal temperature sweep (upon cooling) [20], the ODT of the neat copolymer is determined to be 186 ± 1°C (data not shown here). Addition of FS or CS nanoparticles up to 10 wt% reduces the ODT by as much as ~7°C, depending on surface functionality. This change in ODT is markedly different from that observed in block copolymer nanocomposites composed of a poly(styrene-b-isoprene) diblock copolymer modified with C60 buckyballs [21]. In that case, the ODT is found by dynamic rheology to decrease by 21°C upon incorporation of only 0.04 wt% C60. At higher concentrations of C60 nanoparticles (up to 0.5 wt%), the ODT does not change further, but it does progressively broaden. Such sensitivity is indicative of chemical interactions between the copolymer molecule (specifically, the unsaturated isoprenic units) and the carbonaceous nanoparticles [22], thereby generating a cross-linked material. Such interactions are not expected in the present systems, although we suspect that hydroxyl-terminated silica nanoparticles may bind with the acrylic units upon thermal treatment. To discern the effect of siliceous nanoparticles on mechanical properties at higher concentrations, representative temperature sweeps are presented in Fig. 1 for SM nanocomposites containing 20 wt% of the FS-OH, FS-C8 and CS additives.

Temperature dependence of the dynamic shear moduli (G′, open; G″, filled) for SM nanocomposites containing 20 wt% of three different nanoscale additives: hydroxyl-terminated colloidal silica (CS, circles), hydroxyl-terminated fumed silica (FS-OH, triangles) and octyl-terminated fumed silica (FS-C8, squares). The dotted vertical line identifies an abrupt change in G′ that occurs in the case of the hydroxyl-terminated nanoparticles
These results immediately indicate that the fumed nanoparticles, which exist as branched aggregates commonly measuring on the order of hundreds of nanometers, have a more pronounced effect on the SM copolymer than do CS nanoparticles. Specifically, G′ measured for the nanocomposites with FS consistently exceeds the dynamic loss modulus (G″) over the entire temperature interval examined, even though the copolymer exists as a structureless melt. In contrast, G′ for the CS-containing nanocomposite increases beyond G″ only at low temperatures. Close examination of the data in this figure also reveals that (1) the ODT of the copolymer in the SM/CS nanocomposite persists in the vicinity of 172°C, which constitutes a 14°C reduction in the ODT of the copolymer and (2) a second, less pronounced event appears to occur in the SM/FS nanocomposites at ca. 204°C. This second event is absent in the neat copolymer, as well as in the system modified with CS, whereas neither SM/FS nanocomposite exhibits a discernible ODT. Similar disappearance of the ODT is observed [21] in the case of nanocomposites modified with C60 buckyballs. Over the range from 160 to 200°C, the dynamic moduli measured from the SM-based nanocomposites described in Fig. 1 are well behaved and permit direct assessment of modulus enhancement as functions of nanoparticle concentration and surface chemistry below and above the copolymer ODT.
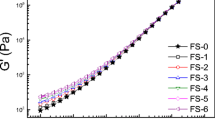
The variation of the dynamic storage modulus (G′) and dissipation factor (tanδ = G″/G′) with nanoparticle concentration is provided at a temperature below the virgin copolymer ODT (at 160°C) in Fig. 2a, 2b, respectively, and above the ODT (at 200°C) in Fig. 2c, 2d, respectively. In Fig. 2a, values of G′ measured from FS-containing nanocomposites generally increase slowly up to ~5 wt% and then increase sharply at higher FS concentrations. In contrast, G′ from the SM/CS nanocomposite remains relatively flat and does not exhibit an abrupt rise until 20 wt% CS, indicating that single CS nanoparticles are less effective at improving the rigidity of the nanocomposite at low loading levels than the aggregated FS nanoparticles. Within the family of FS nanoparticles examined here, the FS-MA variant appears to be the most effective, while the FS-C8 nanoparticles are the least effective, at improving the mechanical properties of the copolymer. This is most likely due to the presumably nonselective nature of the FS-C8 nanoparticles. The FS-MA and FS-OH nanoparticles, on the other hand, can preferentially interact with the PMMA units of the copolymer and therefore immobilize the chains. The solid and dashed lines included in the figure are meant as guides for the eye, but correspond to exponential regressions and fit the data surprisingly well. Similar behavior is observed at 200°C (cf. Fig. 2c) when the copolymer is above its ODT and disordered. It is interesting that the onset of the increase in G′ occurs at about the same concentration of both FS and CS nanoparticles.

Dependence of G′ (a,c) and tanδ (b,d) on nanoparticle concentration for four nanoparticle species—CS (filled circle), FS-OH (open triangle), FS-C8 (open square) and FS-MA (open circle)—at two temperatures (in °C): 160 (a,b) and 200 (c,d). The solid and dashed lines serve as guides for the eye for the FS-MA and CS data, respectively; whereas the shaded region shows the range in G′ over which nanoparticle concentration generally has little effect on nanocomposite mechanical properties. The vertical dotted line identifies the concentration vicinity beyond which the nanocomposites behave solid-like, and the horizontal dotted line (c,d only) signifies where tanδ = 1
In Fig. 2b, tanδ, a direct measure of liquid- versus solid-like behavior, is provided as a function of nanoparticle concentration and shows that, up to the concentration where G′ suddenly increases in Fig. 2a, tanδ is, for the most part, greater than unity. Since tanδ = G″/G′, this observation indicates that the nanocomposite behaves liquid-like. At higher nanoparticle concentrations, tanδ decreases below unity, and the material behaves more solid-like. While there is little systematic variation among the three surface-functionalized FS grades, the SM/CS nanocomposites exhibit the greatest liquid-like tendency, marginally behaving solid-like at 20 wt% CS. As before, similar results are seen in Fig. 2d, which displays tanδ as a function of nanoparticle concentration at 200°C, above the copolymer ODT. Solid-like behavior becomes evident at nanoparticle loading levels that correspond to the sharp rise in G′. The one series that deviates from the data previously discussed with regard to Fig. 2b consists of the SM/FS-C8 nanocomposites. According to Fig. 2d, tanδ for this series attains the highest tanδ value measured (15.5 at 0.1 wt%) in this study and thus exhibits the greatest liquid-like behavior of all the nanocomposites investigated. As the concentration of FS-C8 is increased, however, values of tanδ for this series become comparable to those measured for the other FS, as well as CS, series.
Of all the additives considered here, the discrete CS nanoparticles appear to be the least effective in improving the mechanical properties of the SM copolymer (due to the formation of discrete, rather than interconnected, aggregate structures) and are not considered further. The FS-C8 nanoparticles likewise appear ineffective at low nanoparticle concentrations and temperatures above the copolymer ODT, but their efficacy progressively improves as the nanoparticle concentration increases. Since this series of nanocomposites represents the worst case of FS-based nanoparticles in terms of property-enhancing attributes, it is used to probe the ability of FS nanoparticles to form colloidal networks within the nanostructured copolymer matrix. Figure 3a shows the frequency spectra for G′ and G″ at 140°C and a nanoparticle concentration of 0.5 wt% FS-C8. Viscoelastic behavior is observed wherein G″ exceeds G′ at low ω, but G′ grows larger than G″ at high ω [23]. The crossover point at ωc, where G′ and G″ intersect, yields a characteristic relaxation time (τ) for the material. In this case, τ = 1/ωc is about 1.67 s. At 200°C, similar behavior is observed (data not shown), but τ decreases by an order of magnitude to 0.13 s.

Isothermal frequency spectra acquired for G′ (open circle) and G″ (filled circle) at the following conditions: a 0.5 wt% FS-C8 at 140°C, b 10 wt% FS-C8 at 140°C and c 10 wt% FS-C8 at 200°C
When the concentration of FS-C8 is increased to 10 wt%, the frequency spectra change dramatically from the one displayed in Fig. 3a. In Fig. 3b, G′ is greater than G″ over the entire ω range examined, although both are frequency dependent. These data are reminiscent of weak gel-like materials possessing a long relaxation time [24–27]. In the disordered SM copolymer matrix at 200°C (Fig. 3c), we notice a remarkable behavior: G′ continues to exceed G″, remaining parallel at high ω (with G′ and G″ scaling as ω0.35) but starting to show evidence of a plateau at low ω. The presence of a low frequency plateau in G′ suggests that this material is more gel-like than any of the others [25, 27–29]. Interestingly, the modulus of this sample is lower than that of the specimen portrayed in Fig. 3b. These results taken together suggest the presence of a sample-spanning, self-supporting network within the disordered copolymer melt. In the ordered state, the nanocomposite possesses a higher modulus, but an apparently weaker FS network (due possibly to network disruption upon copolymer ordering). Since this is the least effective modifier of the FS family, it immediately follows that the other additives exhibit comparable, if not more pronounced, network behavior at nanoparticle concentrations of 10 wt% or more.
We now turn our attention to the most promising network-forming nanoparticle grade: FS-MA. Conventional TEM images acquired from relatively thick stained sections of two nanocomposites containing 5 and 20 wt% FS-MA are provided at 0°, 15° and 30° tilt in the top and bottom rows, respectively, of Fig. 4. In the case of the material with 5 wt% FS-MA, the lamellar morphology of the SM copolymer is evident and possesses a period of 20 ± 2 nm. Existence of lamellae confirms that the copolymer molecules are capable of self-organizing (albeit under frustrating conditions) and that an ODT should be observed, which it is according to dynamic rheology [18]. Discrete clusters of FS-MA nanoparticles are likewise visible and measure from ~10 to 35 nm across (which is on the same scale as the primary FS-MA nanoparticles). Recall that, of all the FS nanoparticles investigated here, the FS-MA nanoparticles are expected to be the most uniformly dispersed throughout the copolymer matrix, because they promote the greatest and most consistent property enhancement. Since the nanoparticles reside throughout the specimen and TEM images provide 2D projections of 3D objects, the precise effect of these nanoparticles/clusters on copolymer nanostructure cannot be directly assessed without the use of transmission electron microtomography [30, 31], which will be provided in a forthcoming publication.

TEM images acquired at three different tilt angles (labeled) from SM/FS-MA nanocomposites containing 5 and 20 wt% FS-MA (top and bottom rows, respectively). In each case, the FS-MA nanoparticles appear as electron-opaque (dark) aggregate features, whereas the styrenic lamellae of the SM copolymer are likewise dark due to selective staining. The arrow shows the location of lamellae comprising the copolymer nanostructure, whereas the circled region highlights copolymer lamellae that appear distorted or partially discontinuous
It is apparent, however, from the tilt images corresponding to the nanocomposite with 5 wt% FS-MA that the copolymer lamellae are not highly oriented due, in large part, to the presence of the nanoparticles. Moreover, in several locations throughout the field of view, the lamellae appear distorted or even discontinuous (cf. the circled region at 30° tilt), which is consistent with our previous phase study [18] indicating that the stability of the copolymer nanostructure (discerned from the magnitude of the ODT) in this nanocomposite is lower than that of the neat copolymer. In the case of the nanocomposite containing 20 wt% FS-MA, however, the lamellar nanostructure of the copolymer is altogether eliminated, replaced by a continuous background of nearly constant optical density, whereas the FS-MA nanoparticles form a continuous network that extends throughout the material. These results agree with our findings from dynamic rheology: (1) no ODT is discernible from isochronal temperature sweeps of this nanocomposite and (2) this nanocomposite exhibits solid-like behavior both at low and high temperatures in the melt. Thus, we provide experimental evidence to demonstrate that incorporation of nanoparticles in block copolymer melts can induce sufficient molecular frustration via nanoscale confinement to completely thwart the ability of the copolymer molecules to form a periodic nanostructure.
Conclusions
Addition of native and surface-functionalized siliceous nanoparticles varying in hydrophobicity and inherent aggregation to a nanostructured block copolymer melt has little effect on the rheological properties at low nanoparticle concentrations, but promotes an abrupt increase in G′ and a corresponding decrease in tanδ (below unity) at high nanoparticle loading levels. In this latter regime, the nanocomposite melt behaves solid-like at temperatures above and below the copolymer ODT, suggesting that a colloidal network composed of nanoparticles develops. Existence of such a network is confirmed from mechanical frequency spectra acquired at different nanoparticle concentrations and temperatures. Transmission electron microscopy provides direct visual evidence of a clustered nanoparticle network [32] within the ordered copolymer nanostructure and establishes that two dissimilar nanostructures, both capable of imparting solid-like behavior to soft materials, can coexist in block copolymer nanocomposite melts [33].
References
Hamley IW: The Physics of Block Copolymers. Oxford University Press, New York; 1998.
Lazzari M, Liu G, Lecommandoux S (eds.) (Eds): Block Copolymers in Nanoscience. Wiley, Weinheim; 2006.
Alexandridis P, Lindman B: Amphiphilic Block Copolymers: Self-Assembly and Applications. Elsevier, Amsterdam; 2000.
Alexandridis P, Spontak RJ: Curr. Opin. Colloid Interface Sci. 1999, 4: 130. COI number [1:CAS:528:DyaK1MXlsVOku7o%3D] COI number [1:CAS:528:DyaK1MXlsVOku7o%3D] 10.1016/S1359-0294(99)00022-9
Lodge TP, Pudil B, Hanley KJ: Macromolecules. 2002, 35: 4707. COI number [1:CAS:528:DC%2BD38XjsVyht7w%3D]; Bibcode number [2002MaMol..35.4707L] COI number [1:CAS:528:DC%2BD38XjsVyht7w%3D]; Bibcode number [2002MaMol..35.4707L] 10.1021/ma0200975
Spontak RJ, Patel NP: Developments in Block Copolymer Science and Technology. Edited by: Hamley IW. Wiley, New York; 2004:159–212. 10.1002/0470093943.ch5
Hamley IW: Block Copolymers in Solution: Fundamentals and Applications. Wiley: Hoboken, NJ; 2005.
Spontak RJ, Shankar R, Bowman MK, Krishnan AS, Hamersky MW, Samseth J, Bockstaller MR, Rasmussen KØ: Nano Lett.. 2006, 6: 2115. COI number [1:CAS:528:DC%2BD28XoslalsL0%3D]; Bibcode number [2006NanoL...6.2115S] COI number [1:CAS:528:DC%2BD28XoslalsL0%3D]; Bibcode number [2006NanoL...6.2115S] 10.1021/nl061205u
Tsutsumi K, Funaki Y, Hirokawa Y, Hashimoto T: Langmuir. 1999, 15: 5200. COI number [1:CAS:528:DyaK1MXkt1aqsro%3D] COI number [1:CAS:528:DyaK1MXkt1aqsro%3D] 10.1021/la990246l
Bockstaller MR, Lapetnikov Y, Margel S, Thomas EL: J. Am. Chem. Soc.. 2003, 125: 5276. COI number [1:CAS:528:DC%2BD3sXivVClsbY%3D] COI number [1:CAS:528:DC%2BD3sXivVClsbY%3D] 10.1021/ja034523t
Bockstaller MR, Thomas EL: Phys. Rev. Lett.. 2004, 93: 166106. Bibcode number [2004PhRvL..93p6106B] Bibcode number [2004PhRvL..93p6106B] 10.1103/PhysRevLett.93.166106
Bockstaller MR, Mickiewicz RA, Thomas EL: Adv. Mater.. 2005, 17: 1331. COI number [1:CAS:528:DC%2BD2MXlsVWhsro%3D] COI number [1:CAS:528:DC%2BD2MXlsVWhsro%3D] 10.1002/adma.200500167
Lo C-T, Lee B, Pol VG, Dietz Rago NL, Seifert S, Winans RE, Thiyagarajan P: Macromolecules. 2007, 40: 8302. COI number [1:CAS:528:DC%2BD2sXhtFehu7fO]; Bibcode number [2007MaMol..40.8302L] COI number [1:CAS:528:DC%2BD2sXhtFehu7fO]; Bibcode number [2007MaMol..40.8302L] 10.1021/ma070835v
Kim BJ, Chiu JJ, Yi GR, Pine DJ, Kramer EJ: Adv. Mater.. 2005, 17: 2618. COI number [1:CAS:528:DC%2BD2MXht1Cgt7jL] COI number [1:CAS:528:DC%2BD2MXht1Cgt7jL] 10.1002/adma.200500502
Kim BJ, Fredrickson GH, Hawker CJ, Kramer EJ: Langmuir. 2007, 23: 7804. COI number [1:CAS:528:DC%2BD2sXmtlGgtLc%3D] COI number [1:CAS:528:DC%2BD2sXmtlGgtLc%3D] 10.1021/la700507j
Lee KM, Han CD: Macromolecules. 2003, 36: 804. COI number [1:CAS:528:DC%2BD3sXosVSi]; Bibcode number [2003MaMol..36..804L] COI number [1:CAS:528:DC%2BD3sXosVSi]; Bibcode number [2003MaMol..36..804L] 10.1021/ma020816f
Jain A, Gutmann JS, Garcia CBW, Zhang Y, Tate MW, Gruner SM, Wiesner U: Macromolecules. 2002, 35: 4862. COI number [1:CAS:528:DC%2BD38XjvFagtLg%3D]; Bibcode number [2002MaMol..35.4862J] COI number [1:CAS:528:DC%2BD38XjvFagtLg%3D]; Bibcode number [2002MaMol..35.4862J] 10.1021/ma025511f
Gaines MK, Smith SD, Samseth J, Bockstaller MR, Thompson RB, Rasmussen KØ, Spontak RJ: Soft Matter. 2008, 4: 1609. COI number [1:CAS:528:DC%2BD1cXoslentL4%3D] COI number [1:CAS:528:DC%2BD1cXoslentL4%3D] 10.1039/b805540h
Zheng QXS, Braunfeld MB, Sedat JW, Agard DA: J. Struct. Biol.. 2004, 147: 91. 10.1016/j.jsb.2004.02.005
Rosedale JH, Bates FS: Macromolecules. 1990, 23: 2329. COI number [1:CAS:528:DyaK3cXhsF2gtro%3D]; Bibcode number [1990MaMol..23.2329R] COI number [1:CAS:528:DyaK3cXhsF2gtro%3D]; Bibcode number [1990MaMol..23.2329R] 10.1021/ma00210a032
Zhao Y, Hashimoto T, Douglas JF: J. Chem. Phys.. 2009, 130: 124901. Bibcode number [2009JChPh.130l4901Z] Bibcode number [2009JChPh.130l4901Z] 10.1063/1.3089667
Laiho A, Ras RHA, Valkama S, Ruokolainen J, Österbacka R, Ikkala O: Macromolecules. 2006, 39: 7648. COI number [1:CAS:528:DC%2BD28XhtVShs7zJ]; Bibcode number [2006MaMol..39.7648L] COI number [1:CAS:528:DC%2BD28XhtVShs7zJ]; Bibcode number [2006MaMol..39.7648L] 10.1021/ma061165g
Dealy JM, Larson RG: Structure and Rheology of Molten Polymers: From Structure to Flow Behavior and Back Again. Hanser, Munich; 2006.
English RJ, Raghavan SR, Jenkins RD, Khan SA: J. Rheol.. 1999, 43: 1175. COI number [1:CAS:528:DyaK1MXlt1Cru78%3D]; Bibcode number [1999JRheo..43.1175E] COI number [1:CAS:528:DyaK1MXlt1Cru78%3D]; Bibcode number [1999JRheo..43.1175E] 10.1122/1.551026
Tayal A, Pai VB, Khan SA: Macromolecules. 1999, 32: 5567. COI number [1:CAS:528:DyaK1MXkvFWntbY%3D]; Bibcode number [1999MaMol..32.5567T] COI number [1:CAS:528:DyaK1MXkvFWntbY%3D]; Bibcode number [1999MaMol..32.5567T] 10.1021/ma990167g
Chiou BS, Raghavan SR, Khan SA: Macromolecules. 2001, 34: 4526. COI number [1:CAS:528:DC%2BD3MXjs1Sns7Y%3D]; Bibcode number [2001MaMol..34.4526C] COI number [1:CAS:528:DC%2BD3MXjs1Sns7Y%3D]; Bibcode number [2001MaMol..34.4526C] 10.1021/ma010281a
Kumar R, Kalur GC, Ziserman L, Raghavan SR: Langmuir. 2007, 23: 12849. COI number [1:CAS:528:DC%2BD2sXhtlSkurnI] COI number [1:CAS:528:DC%2BD2sXhtlSkurnI] 10.1021/la7028559
English RJ, Laurer JH, Spontak RJ, Khan SA: Ind. Eng. Chem. Res.. 2002, 41: 6425. COI number [1:CAS:528:DC%2BD38XoslShs7Y%3D] COI number [1:CAS:528:DC%2BD38XoslShs7Y%3D] 10.1021/ie020409s
Pai VB, Khan SA: Carbohydr. Polym.. 2002, 49: 207. COI number [1:CAS:528:DC%2BD38XjtVSit7o%3D] COI number [1:CAS:528:DC%2BD38XjtVSit7o%3D] 10.1016/S0144-8617(01)00328-9
Jinnai H, Spontak RJ, Nishi T: Macromolecules. 2010, 43: 1675. COI number [1:CAS:528:DC%2BC3cXht1KgtL0%3D]; Bibcode number [2010MaMol..43.1675J] COI number [1:CAS:528:DC%2BC3cXht1KgtL0%3D]; Bibcode number [2010MaMol..43.1675J] 10.1021/ma902035p
Abetz V, Spontak RJ, Talmon Y: Macromolecular Engineering Precise Synthesis, Materials Properties, Applications. Edited by: ed. by Matyjaszewski K, Gnanou Y, Leibler L. Wiley, Weinheim; (2007):1649–1685.
Rahedi AJ, Douglas JF, Starr FW: J. Chem. Phys.. 2008, 128: 024902. Bibcode number [2008JChPh.128b4902R] Bibcode number [2008JChPh.128b4902R] 10.1063/1.2815809
Thompson RB, Ginzburg VV, Matsen MW, Balazs AC: Science. 2001, 292: 2469. COI number [1:CAS:528:DC%2BD3MXkvFWmurc%3D] COI number [1:CAS:528:DC%2BD3MXkvFWmurc%3D] 10.1126/science.1060585
Acknowledgments
This work was supported by the Research Council of Norway under the NANOMAT Program. M. K. G. expresses her gratitude for a GEM Fellowship and a NOBCChE Procter & Gamble Fellowship.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Gaines, M.K., Smith, S.D., Samseth, J. et al. Nanoparticle Network Formation in Nanostructured and Disordered Block Copolymer Matrices. Nanoscale Res Lett 5, 1712 (2010). https://doi.org/10.1007/s11671-010-9775-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11671-010-9775-y