
Abstract
We present first-principles density-functional calculations for the structural, electronic, and magnetic properties of substitutional 3d transition metal (TM) impurities in two-dimensional black and blue phosphorenes. We find that the magnetic properties of such substitutional impurities can be understood in terms of a simple model based on the Hund’s rule. The TM-doped black phosphorenes with Ti, V, Cr, Mn, Fe, and Ni impurities show dilute magnetic semiconductor (DMS) properties while those with Sc and Co impurities show nonmagnetic properties. On the other hand, the TM-doped blue phosphorenes with V, Cr, Mn, and Fe impurities show DMS properties, with Ni impurity showing half-metal properties, whereas Sc- and Co-doped systems show nonmagnetic properties. We identify two different regimes depending on the occupation of the hybridized electronic states of TM and phosphorous atoms: (i) bonding states are completely empty or filled for Sc- and Co-doped black and blue phosphorenes, leading to nonmagnetic; (ii) non-bonding d states are partially occupied for Ti-, V-, Cr-, Mn-, Fe- and Ni-doped black and blue phosphorenes, giving rise to large and localized spin moments. These results provide a new route for the potential applications of dilute magnetic semiconductor and half-metal in spintronic devices by employing black and blue phosphorenes.
PACS numbers: 73.22.-f, 75.50.Pp, 75.75. + a
Similar content being viewed by others


Background
Two-dimensional (2D) materials, graphene and silicene, are currently the subject of intense theoretical and experimental research especially for their novel electronic device applications [1, 2]. Graphene and silicene have demonstrated many exquisite phenomena originating from the characteristic conical dispersion and chiral behavior of their valence and conduction bands around the Fermi level [3, 4]. Generally speaking, the nanostructures of graphene and silicene such as nanoribbons, nanotubes, and their interconnections have opened new routes for experimental and theoretical studies in the field of nanoelectronics [5]. Very recently, black phosphorene, a single layer of black phosphorus (BP), was successfully fabricated through exfoliation from the bulk black phosphorus [6] and therefore becomes, besides graphene and silicene, another stable elemental 2D material. The black phosphorene presents some advantages superior to other previously studied 2D semiconductors because of its intriguing electronic properties, thereby drawing enormous interest from the society of materials science [7–13]. Recently, Li et al. [14] reported that black phosphorene could be applied to the channel of the field-effect transistor (FET) device that has a high carrier mobility of ∼ 103cm2/V ·s and an on/off ratio of ∼ 104 at room temperature. As the allotrope of black phosphorene, blue phosphorene has the same stability as black phosphorene at room temperature, and its band gap is larger than black phosphorene [15]. These good electronic properties of black and blue phosphorene nanosheets can be useful for the development of future nanoelectronic devices, spintronics, and related applications [16–23].
For the design of practical electronic devices, defects and impurities have been employed to tune the electrical, optical, and other properties. Over the last decades, the resulting dilute magnetic semiconductors (DMS) and half-metals have achieved important developments both in fundamental aspects and prospective technological applications [24, 25]. Indeed, it was possible to understand the underlying mechanisms of interaction between dilute magnetic impurities allowing ferromagnetic semiconductors at room temperature. For prospective applications, the integration between 2D semiconductors and magnetic data storage enables the development of two-dimensional spintronic devices such as spin valve, spin-based transistors, non-volatile magnetoresistive memories, and even magnetically enhanced optoelectronics devices.
Meanwhile, in spite of the success of 2D materials such as graphene, silicene, transition metal dichalcogenides (TMDCs) [26], and black phosphorene [17, 27, 28], there has been rare study on the dilute magnetic characters of doped 2D black phosphorene except the work of Hashmi et al. [29] and Sui et al. [30], while half-metal properties in doped blue phosphorene have remained unexplored so far. From a technical point of view, 2D semiconductors have other superior factors that can be exploited in magnetic or spintronic devices. First, the carrier concentration can be externally controlled by voltage gating. Secondly, there is room to improve the control of the impurity concentration, for example, by employing adatoms as impurities with concentrations above the solubility limit. In practice, studies of magnetic semiconductor nanostructures with lower dimensionalities, including semiconductor nanocrystals and nanowires doped with transition metals (TM), demonstrated that the confinement effect and the improved control of magnetic dopants can be used to increase the Curie temperature [31].
In this work, we focus on substitutional 3d TM impurities (from Sc to Ni) in black and blue phosphorenes to investigate their dilute magnetic characters and half-metal properties. Using first-principles density functional theory (DFT) calculations, we study the structural, electronic, and magnetic properties of substitutional 3d TM impurities in black and blue phosphorenes. One of our key results is that the electronic and magnetic properties of these substitutional impurities can be understood by a simple model based on the hybridization between the TM d orbitals and the defect (i.e., phosphorous vacancy) levels. This model together with the calculated band structure provides an explanation for the non-trivial behaviors of the binding energy and the spin moments for all the systems considered. Concisely, we distinguish two different regimes that depend on the electron filling of TM-phosphorous hybridized levels: (i) completely unoccupied (occupied) bonding states for Sc (Co) lead to nonmagnetic and (ii) partially occupied non-bonding d shell for Ti, V, Cr, Mn, Fe, and Ni give rise to large and localized spin moments.
This paper is organized as follows. After a brief description of the computational details, we present the geometry structures, binding energies, and magnetic properties of all the substitutional TM impurities studied. We also present the general ideas behind our model of the metal-phosphorus hybridization in the considered systems. Then, the electronic structure of the unreconstructed C\(_{2h}^{3}\) phosphorus vacancy in pristine phosphorene, along with the electronic structures of the different groups of impurities, are presented. Finally, we give a summary with some general conclusions.
Methods
The present DFT calculations were performed using the Vienna Ab initio Simulation Package (VASP) code with a plane-wave basis set [32]. Projector augmented wave (PAW) potentials [33] were used to describe the core electrons, and the generalized gradient approximation (GGA) of Perdew, Burke, and Ernzernhof (PBE) [34] was adopted for exchange-correlation energy. To examine the reliability of the PBE method on the magnetic properties of the black and blue phosphorenes containing 3d TM impurity atoms, we also considered the effect of the on-site Coulomb interaction U on the magnetic property within the PBE + U method. The values of U are 4.0, 5.5, 3.3, 3.5, 4.3, 3.3, and 6.5 eV for Sc-, Ti-, V-, Cr-, Mn-, Fe-, Co-, and Ni-doped systems, respectively. The kinetic energy cutoff of the plane-wave basis set used was 500 eV, and for the structural optimization, convergence of Hellmann-Feynman residual forces less than 0.01 eV/Å per atom was achieved. Because the convergence with respect to the number of k-points was especially critical to obtain accurate results for the spin moment in the systems studied, we used an adequate number of k-points for all the different supercell sizes, equivalent to 9 × 9 × 1 Monkhorst-Pack [35] sampling. The Fermi level was smeared by the Gaussian method with a width of 0.05 eV. Most of our results were obtained using 2 × 2/4 × 4 crystallographic symmetrical supercells of black and blue phosphorenes, with a doping concentration of 3.13 %, as shown in Fig. 1 a, b. We also checked the results by performing calculations using larger supercells up to 4 × 4/6 × 6 [concentration of 1.56(1.39) %] for several elements. In order to avoid spurious interactions between periodic images of the defective phosphorene layer, a vacuum spacing perpendicular to the plane was employed to be larger than ∼15 Å.

a Top and side views of a diamond-like 2 × 2 supercell of black phosphorene used in the present calculations. b Top and side views of blue phosphorene. c Spin density of black phosphorene containing a vacancy with an isosurface of 0.25 e/Å3
Results and Discussion
Geometrical Parameters and Binding Energies
The typical structure of the systems studied in this paper is presented in Fig. 1. Figure 1 a shows the diamond-like 2 × 2 supercell structure of monolayer black phosphorene, with a doping concentration of 3.13 %. In view of symmetry and doping isotropy, the diamond-shape unit cell was employed instead of the rectangle unit cell. As shown in Fig. 1 a, the optimized lattice constants are a 1=3.310 Å, a 2=4.589 Å, and \(a_{1}^{\prime }=a_{2}^{\prime }=\left ({a_{1}^{2}}+{a_{2}^{2}}\right)^{1/2}=5.658\) Å, and the angle between the basis vectors \(a_{1}^{\prime }\) and \(a_{2}^{\prime }\) is 71.89°. These values are consistent with experiment and other theoretical calculations [36]. Figure 1 b displays the optimized structure of the blue phosphorene with lattice constants \(a_{1}^{\prime \prime }=a_{2}^{\prime \prime }=3.330\) Å, and their angle θ ′=60°, which are in good agreement with previous DFT calculations [15].
We begin to study pure black and blue phosphorenes with a monovacancy. Because the blue phosphorene with monovacancy has no magnetism, we only plot the spin charge density of the black phosphorene with monovacancy in Fig. 1 c. Figure 1 c shows the spin density of black phosphorene with a monovacancy. Similar to the carbon atoms around the vacancy in graphene, the phosphorus atoms around the vacancy in phosphorene undergo a Jahn-Teller distortion, and two of the phosphorus atoms close to the vacancy site move towards each other to form a P-P distance of 1.832 Å, which is 0.408 Å smaller than that of the intrinsic phosphorene. The ground state of the system has a magnetic moment of 1.00 μB/unit cell, most of which is concentrated at the two P atoms with the unsaturated bonds, as seen in Fig. 1 c.
The structural parameters and energetic properties of the substitutional TMs in black and blue phosphorenes are shown in Fig. 2. For TM-doped black phosphorene, the bond angles θ 1 and θ 2 monotonically increase from Sc to Ni (see Fig. 2 b). Meanwhile, the bond lengths d 1 and d 2 decrease for Sc-, Ti-, and V-doped systems and increase for Cr-doped system, decrease again for Mn-, Fe-, and Co-doped systems, and then increase for Ni-doped system (see Fig. 2 a). These behaviors of the bond lengths and bond angles reflect the size of the TM atoms. As for TM-doped blue phosphorene, the bond length d 3 decreases from Sc to Mn and then increases from Mn to Ni (see Fig. 2 d), while the band angle θ 3 shows an oscillating behavior (see Fig. 2 e).

a, b Structural parameters and c binding energies of the substitutional TM-doped black phosphorenes. The corresponding ones of the substitutional TM-doped blue phosphorenes are given in d, e, and f
Figure 2 c shows the calculated binding energies (E b) of the TM-doped black phosphorenes, where E b is defined as –(E total– E phosphorene– E atom). Here, E total is the energy of the whole configuration, E phosphorene is the energy of the phosphorene with a vacancy, and E atom represents the energy of an isolated dopant atom. We find a continuous increase of the binding energy from Sc to Cr, and then decrease from Mn to Ni, and the binding energies for the considered TMs are in the range of 0.375–5.466 eV. Interestingly, Cr-doped system has the maximum binding energy. This peculiar behavior is related with the interplay between the energy down-shift and the compression of the 3d shell of the TM as the atomic number increases. Although this explanation will be more clear when the metal-phosphorus hybridization levels are discussed below, we note that the behavior of the binding energies of the substitutional 3d TM arises from two competing effects:
-
(i)
From Sc to Cr, the decrease of d 1 and d 2 reflects an increase in the bonding strength between the TM and phosphorous atoms
-
(ii)
From Mn to Ni, as the 3d shell is occupied, its hybridization with the phosphorous vacancy states is weakened to decrease the binding energy
It is noticeable that this trend of the energetics for the TM-doped black phosphorenes is very similar to that for the TM-doped blue phosphorenes (see Fig. 2 f).
Spin Moments
The spin moments of substitutional TMs in black and blue phosphorenes are displayed in Fig. 3 a, together with those of the isolated TM atoms. We find that the spin moments of the isolated TM atoms are 1, 3, 5, 6, 5, 4, 3, and 2 μB from Sc to Ni, respectively. On the other hand, the TM-substituted black phosphorenes have the zero magnetic moment for Sc and Co, but 1, 2, 3, 2, 1, and 1 μB for Ti, V, Cr, Mn, Fe, and Ni, respectively, which are the same as the corresponding cases in blue phosphorene. For both of the TM-substituted black and blue phosphorenes, we analyze the charge transfer using Bader charge (see Table 1). We find that for both cases, the TM atoms lose electron charges while the nearest phosphorous atoms gain electron charges. It is notable that the magnitudes of gained and lost charges decrease as the atomic number increases.

a Spin moments of the isolated TMs and their substitutions in black and blue phosphorenes as a function of the number of valence electrons (Slater-Pauling-type plot). Schematic diagram of spin moment in doped black phosphorene b and blue phosphorene c in terms of Hund’s rule, respectively
Interestingly, as shown in Fig. 3 a, the total spin moments have the integer values of 0, 1, 2, 3, 2, 1, 0, and 1 μB for Sc-, Ti-, V-, Cr-, Mn-, Fe-, Co-, and Ni-doped black and blue phosphorenes, respectively. According to a first-principles study of substitutional TM impurities in graphene, the spin moments are calculated to be 0, 0, 1, 2, 3, 2, 1, and 1 μB for Sc-, Ti-, V-, Cr-, Mn-, Fe-, Co-, and Ni-doped graphene systems, respectively. These values are well compared with 0, 1, 2, 3, 2, 1, 0, and 1 μB for Sc-, Ti-, V-, Cr-, Mn-, Fe-, Co-, and Ni-doped black and blue phosphorenes, respectively. It is interesting to note that the spin moment of each TM impurity (except Sc and Ni) in graphene is smaller by 1 μB compared to the corresponding one in black and blue phosphorenes. This may be attributed to the different bonding natures of graphene and phosphorene: i.e., sp 2 bonding in graphene and sp 3 bonding in phosphorene. Since one valence electron of TM impurities in graphene participates in π bonding with neighboring C atoms, the spin moment is likely to decrease by 1 μB. To understand this deeply, we draw the schematic diagram of spin moment according to Hund’s rule in Fig. 3 b. We here note that the valence electron configurations of Sc, Ti, V, Cr, Mn, Fe, Co, and Ni are 3 d 14s 2, 3 d 24s 2, 3 d 34s 2, 3 d 54s 1, 3 d 54s 2, 3 d 64s 2, 3 d 74s 2, 3 d 84s 2, respectively. Briefly, we can distinguish the several regimes depending on the filling of electronic levels:
-
■■■
(i) Sc-doped black phosphorene have the empty Sc-phosphorous bonding levels, leading to a zero spin moment.
-
■■■
(ii) Co-doped black phosphorene have fully occupied Co-phosphorous bonding levels, leading to a zero spin moment.
-
■■■
(iii) Ti-, V-, and Cr-doped black phosphorene have partially occupied TM-phosphorous bonding levels with the spin moments of 1.00, 2.00, and 3.00 μB, respectively.
-
■■■
(iv) Mn-, Fe-, and Ni-doped black phosphorene have partially occupied non-bonding 3d levels with the spin moments of 2.00, 1.00, and 1.00 μB.
It is notable that the spin moments of TM-doped blue phosphorene (see Fig. 3 c) are the same as those of black phosphorene because the energy states of s and d in the outermost orbital of TM atoms and phosphorus atom are very close to each other.
To explore the underlying mechanism of the magnetic moments in TM-doped black and blue phosphorenes, the Mulliken population analysis was performed to list the results in Table 2. We find that the spin moments of the TM impurities (S M) have a dominant contribution for the nearest phosphorus neighbors (S P1 and S P2). The calculated spin moments of TM impurities for Ti, V, Cr, Mn, Fe, and Ni in doped black phosphorene are S M=0.986,1.977,3.082,2.207,1.097, and 0.953 μB, respectively, close to the above-discussed Hund’s analysis. Similarly, the spin moments of TM impurities for Ti, V, Cr, Mn, Fe and Ni in doped blue phosphorenes are S M=0.992,2.032,3.147,1.954,1.247 and, 0.967 μB, respectively.
Analysis of the Electronic Structures
Before the analysis of the electronic structures of TM-doped black and blue phosphorene, we first examine the electronic structure of a single phosphorous vacancy in black phosphorene. As substitutional impurities in black phosphorene, most of the TM atoms studied here exhibit a symmetrical configuration of C\(_{2h}^{3}\) [37]. For this reason, it is particularly instructive to analyze their electronic structures with the hybridization between the atomic levels of the TM atoms and those associated with a relaxed C\(_{2h}^{3}\) symmetrical phosphorus vacancy. As shown in Fig. 4, the C\(_{2h}^{3}\) vacancy shows a considerable spin polarization of 1.00 μB, indicating a dilute magnetic property.

(Color online). Band structure and density of states of the undoped defective black phosphorene. The red lines represent majority spin band, while the black lines represent minority spin band. The energy zero represents the Fermi level
To further shed light on the underlying mechanism of magnetic properties in the TM-doped black and blue phosphorene structures, we plot the spin-polarized band structures of TM-doped black and blue phosphorenes in Fig. 5 a, b, respectively. Interestingly, the majority and minority spin bands for Ti-, V-, Cr-, Mn-, Fe-, and Ni-doped black phosphorene show semiconducting characters (see Fig. 5 a), indicating dilute magnetic properties. On the other hand, Sc- and Co-doped black phosphorene exhibit zero spin moment, whereas Ti- and Ni-doped blue phosphorene show half-metal characters (see Fig. 5 b). Note that V-, Cr-, Mn-, and Fe-doped blue phosphorenes exhibit dilute semiconducting characters, while Sc- and Co-doped blue phosphorenes have zero spin moment.

(Color online). Band structures of Sc-, Ti-, V-, Cr-, Mn-, Fe-, Co- and Ni-doped black (a) and blue (b) phosphorenes, respectively. The red (black) lines represent the majority (minority) spin band. The energy zero represents the Fermi level
Generally, substitutional TM impurities in black and blue phosphorenes exhibit very similar behaviors in their energetic and magnetic properties. This result indicates that the structural differences of black- and blue-phosphorene lattices are insensitive to determine the energetic and magnetic properties of TM-doped black and blue phosphorenes, as shown in Figs. 2, 3, and 5.
It is interesting to examine the effect of the on-site Coulomb interaction U on the magnetic properties of the substitutional 3d TM impurities in black and blue phosphorenes. We perform the PBE + U calculations for all the considered systems, where the values of U = 4.0, 5.5, 3.3, 3.5, 3.5, 4.3, 3.3, and 6.5 eV are chosen for Sc-, Ti-, V-, Cr-, Mn-, Fe-, Co-, and Ni-doped systems, respectively [38–40]. The calculated PBE + U band gaps (Eg) of Sc-, Ti-, V-, Cr-, Mn-, Fe-, Co-, and Ni-doped black and blue phosphorenes are listed in Table 2. We find that Eg of the magnetic semiconductor obtained using PBE + U increases by ∼30 % compared to the PBE results. However, it is noticeable that the spin moment does not change depending on the PBE and PBE + U methods. Interestingly, we find that the PBE + U band structure of Ti-doped blue phosphorene shows a magnetic semiconductor property with a gap opening (see Fig. 6), different from the half-metallic character predicted by PBE. This indicates that U in Ti-doped blue phosphorene splits the narrow half-filled bands crossing the Fermi level into lower and upper Hubbard bands. On the other hand, the half-metallic character of Ni-doped blue phosphorene predicted by PBE is preserved in the PBE + U band structure (see Fig. 6), because the bands crossing the Fermi level have relatively larger band widths compared to those in Ti-doped blue phosphorene (see Fig. 5 b).

(Color online). Band structures of Ti- and Ni-doped blue phosphorenes obtained using the PBE + U calculation with U = 5.5, 6.5 eV, respectively. The red (black) lines represent the majority (minority) spin band. The energy zero represents the Fermi level
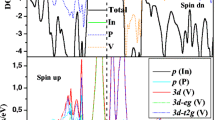
A general picture of the dilute magnetic and half-metal features of the TM-doped black and blue phosphorenes can be seen from the analysis of the spin-polarized total and partial DOS, as shown in Fig. 7. As for Sc- and Co-doped black phosphorenes, the total DOS of the majority and minority states are completely compensated with each other, yielding zero spin moment (see Fig. 7 a). It is found that for Ti-, V-, Cr-, Mn-, Fe- and Ni-doped black phosphorenes, the total DOS of the majority and minority states are not compensated below E F and show a gap opening, indicating dilute magnetic semiconducting properties. We note that the DOS of Sc- and Co-doped blue phosphorene show nonmagnetic properties; those of Ti- and Ni-doped blue phosphorene show half-metal properties; V-, Cr-, Mn-, and Fe-doped blue phosphorenes show dilute magnetic semiconductor characters (see Fig. 7 b). From the analysis of the spin-polarized total and partial DOS, it is seen that the magnetic moments are well localized at the TM atom site, and the d xy and \(d_{x^{2}-y^{2}\phantom {\dot {i}\!}}\) orbitals are dominant for the contribution to the partial DOS.

(Color online). Spin polarized total (upper panel) and partial (lower panel) density of states of Sc-, Ti-, V-, Cr-, Mn-, Fe-, Co-, and Ni-doped black (a) and blue (b) phosphorenes, respectively. The energy zero represents the Fermi level
Conclusions
We have performed a first-principles DFT calculation for the structural, energetic, and magnetic properties of a series of substitutional 3d TM impurities in black and blue phosphorenes. We provided a simple model based on Hund’s rule for understanding the calculated electronic and magnetic properties of the considered systems, where the dilute-semiconducting and half-metal features, spin moment, and binding energy are varied depending on the atomic number of the TM atoms. The spin-polarized band structures and DOS calculations show that for black phosphorene, the Ti-, V-, Cr-, Mn-, Fe-, and Ni-doped systems have dilute magnetic semiconductor properties, while Sc- and Co-doped systems have no magnetism. For blue phosphorene, the Ni-doped system shows half-metal properties, while V-, Cr-, Mn-, and Fe-doped systems show dilute magnetic semiconductor characters, Sc- and Co-doped systems show non-magnetism.
Since substitutional impurities of 3d TM atoms in black and blue phosphorenes exhibit some intriguing electronic and magnetic properties, such doped systems can provide an interesting route to tune various functions for spin electronic devices based on black and blue phosphorenes. This functional ability together with the high stability of substitutional impurities can open a route to fabricate ordered arrays of these impurities at predefined locations, which would allow the experimental tests of the theoretical predictions of unusual magnetic interactions mediated by black and blue phosphorenes.
References
Geim AK, Grigorieva IV (2013) Van der Waals heterostructures. Nature 499: 419.
Vogt P, Padova PD, Quaresima C, Avila J, Frantzeskakis E, Asensio MC, Resta A, Ealet B, Lay GL (2012) Silicene: compelling experimental evidence for graphenelike two-dimensional silicon. Phy Rev Lett 108: 155501.
Bianco E, Butler S, Jiang S, Restrepo OD, Windl W, Goldberger E (2013) Stability and exfoliation of germanane: a germanium graphane analogue. ACS Nano 7: 4414.
Butler SZ, Hollen SM, Cao L, Cui Y, Gupta JA, et al (2013) Progress, challenges, and opportunities in two-dimensional materials beyond graphene. ACS Nano 7: 2898.
Zhang S, Li C, Li SF, Sun Q, Guo ZX, Jia Y (2014) Intrinsic spin dependent and ferromagnetic stability on edge saturated zigzag graphene-like carbon-nitride nanoribbons. Appl Phys Lett 104: 172111.
Reich ES (2014) Phosphorene excites materials scientists. Nature 506: 19.
Zhu ZL, Li C, Yu WY, Chang DH, Sun Q, Jia Y (2014) Magnetism of zigzag edge phosphorene nanoribbons. Appl Phys Lett 105: 113105.
Tran V, Soklaski R, Liang YF, Yang L (2014) Layercontrolled band gap and anisotropic excitons in few-layer black phosphorus. Phys Rev B Phys 89: 235319.
Fei RX, Yang L (2014) Strain-engineering the anisotropic electrical conductance of few-layer black phosphorus. Nano Lett 14: 2884.
Fei RX, Yang L (2014) Lattice vibrational modes and Raman scattering spectra of strained phosphorene. Appl Phys Lett 105: 083120.
Tranm V, Yang L (2014) Unusual scaling laws of the band gap and optical absorption of phosphorene nanoribbons. Phys Rev B 89: 245407.
Zhu ZL, Yu WY, Ren XY, Sun Q, Jia Y (2015) Grain boundary in phosphorene and its unique roles on C and O doping. Europ Lett 109: 47003.
Yu WY, Zhu ZL, Niu CY, Li C, Cho JH, Jia Y (2015) Anomalous doping effect in black phosphorene using first-principles calculations. Phys Chem Chem Phys 17: 16351.
Li L, Yu Y, Ye GJ, Ge Q, Ou X, Wu H, Feng D, Chen XH, Zhang YB (2014) Black phosphorus fieled-effect transistors. Nat Nanotech 9: 372.
Zhu Z, Tomanek D (2014) Semiconducting layered blue phosphorus: a computational study. Phys Rev Lett 112: 176802.
Buscema M, Groenendijk DJ, Blanter SI, Steele GA, Van der Zant HSJ, Gomez AC (2014) Fast and broadband photoresponse of few-layer black phosphorus field-effect transistors. Nano Lett 14: 3347.
Cai YQ, Ke QQ, Zhang G, Zhang YW (2015) Energetics, charge transfer, and magnetism of small molecules physisorbed on phosphorene. J Phys Chem C 119: 3102.
Kulish VV, Malyi OI, Perssonc C, Wu P (2015) Adsorption of metal adatoms on single-layer phosphorene. Phys Chem Chem Phys 17: 992.
Kulish VV, Malyi OI, Persson C, Wu P (2015) Phosphorene as an anode material for Na-ion batteries: a first-principles study. Phys Chem Chem Phys 17: 13921.
Boukhvalov DW, Rudenko AN, Prishchenko DA, Mazurenko VG, Katsnelson MI (2015) Chemical modifications and stability of phosphorene with impurities: a first principles study. Phys Chem Chem Phys 17: 15209.
Qin GZ, Yan QB, Qin ZZ, Yue SY, Hu M, Su G (2015) Anisotropic intrinsic lattice thermal conductivity of phosphorene from first principles. Phys Chem Chem Phys 17: 4854.
Liu X, Wen YW, Chen ZZ, Shan B, Chen R (2015) A first-principles study of sodium adsorption and diffusion on phosphorene. Phys Chem Chem Phys 17: 16398.
Zheng HL, Zhang JM, Yang BS, Du XB, Yan Y (2015) A first-principles study on the magnetic properties of nonmetal atom doped phosphorene monolayers. Phys Chem Chem Phys 17: 16341.
Zhu ZL, Chen WG, Sun Q, Jia Y (2014) Half-metal behaviour mediated by self-doping of topological line defect combining with adsorption of 3d transition-metal atomic chains in graphene. J Phys D 47: 055303.
Feng Y, Ji WX, Huang BJ, Chen XL, Li F, Li P, Zhang CW, Wang PJ (2015) The magnetic and optical properties of 3d transition metal doped SnO2 nanosheets. RSC Adv 5: 24306.
Wang QH, Kalantar-Zadeh K, Kis A, Coleman JN, Strano MS (2012) Low frequency electronic Noise in single-layer MoS2 transistors. Nat Nanotechnol 7: 699.
Hu T, Hong JS (2015) First-principles ptudy of metal adatom adsorption on black phosphorene. J Phys Chem C 119: 8199.
Khan I, Hong JS (2015) Manipulation of magnetic state in phosphorene layer by non-magnetic impurity doping. New J Phys 17: 023056.
Hashmi A, Hong J (2015) Transition metal doped phosphorene: first-principles study. J Phys Chem C 119: 9198.
Sui XL, Si C, Shao B, Zou XL, Wu J, Gu BL, Duan WH (2015) Tunable magnetism in transition-metal-decorated phosphorene. J Phys Chem C 119: 10059.
Seixas L, Carvalho A, Castro Neto AH (2015) Atomically thin dilute magnetism in Co-doped phosphorene. Phys Rev B 91: 155138.
Kresse G, Furthmller J (1996) Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput Mater Sci 6: 15.
Blochl PE (1994) Projector augmented-wave method. Phys Rev B 50: 17953.
Perdew JP, Burke K, Ernzerhof M (1996) Generalized gradient approximation made simple. Phys Rev Lett 77: 3865.
Monkhosrt HJ, Pack JD (1976) Special points for Brillouin-zone integrations. Phys Rev B 13: 5188.
Qiao JS, Kong XH, Hu ZX, Yang F, Ji W (2014) High-mobility transport anisotropy and linear dichroism in few-layer black phosphorus. Nat Comm 5: 4475.
Ribeiro SJ, Almeida RM, Cancado LG, Dresselhaus MS, Jorio A (2015) Group theory for structural analysis and lattice vibrations in phosphorene systems. Phys Rev B 91: 205421.
Rai DP, Thapa RK (2013) An ab initio study of the half metallic properties of Co2TGe (T = Sc, Ti, V, Cr, Mn, Fe): LSDA + U method. J Korean Phys Soc 62: 1652.
Huang WL, Zhu Q, Ge W, Li H (2011) Oxygen-vacancy formation in LaMO3 (M = Ti, V, Cr, Mn, Fe, Co, Ni) calculated at both GGA and GGA + U levels. Comput Mater Sci 50: 1088.
Pari G, Mathi Jaya S, Subramoniam G, Asokamani R (1995) Density-functional descriytion of the electronic structure of LaMO3 (M = Sc, Ti, V, Cr, Mn, Fe, Co, Ni). Phys Rev B 51: 16575.
Acknowledgements
We thank Prof. Zhenyu Zhang for the helpful discussions. This work was supported by the National Basic Research Program of China (Grant No. 2012CB921300), the National Natural Science Foundation of China (Grant Nos. 11504332, 11274280 and 11304288), and the National Research Foundation of Korea (Grant No. 2015R1A2A2A01003248).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
YJ and J-HC conceived the idea. WY performed the calculations, analyzed the data, and wrote the manuscript. ZZ, C-YN and CL also participated in the discussions of the calculated results. All the authors have read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License(http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Yu, W., Zhu, Z., Niu, CY. et al. Dilute Magnetic Semiconductor and Half-Metal Behaviors in 3d Transition-Metal Doped Black and Blue Phosphorenes: A First-Principles Study. Nanoscale Res Lett 11, 77 (2016). https://doi.org/10.1186/s11671-016-1296-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s11671-016-1296-x