
Abstract
The present paper is devoted to the problem of sintering ceramics based on yttria-stabilized zirconia (Y-TZP). In this paper, we studied the effect of two obtaining methods (co-precipitation and technical hydrolysis) on sintering kinetics of Y-TZP nanopowders. We used the constant rate of heating (CRH) method at different heating rates for determining the sintering mechanisms. The basic mechanism and activation energy (Q) of diffusion at the initial sintering stage were estimated using the sintering rate equations that are applicable to the CRH data. We found that nanopowder 3Y-TZP produced by the co-precipitation method (DIPE) was sintered according to the volume diffusion mechanism (n = 1/2) and nanopowder TZ-3Y (TOSOH) produced by the technical hydrolysis was sintered according to the grain boundary diffusion mechanism (n = 1/3).
Similar content being viewed by others

Background
Yttria-stabilized zirconia (Y-TZP) ceramics has excellent mechanical properties, such as high fracture toughness, strength, and hardness. This ceramic material is widely used for different applications [1]. The properties of Y-TZP ceramics strongly depend on the obtaining conditions of nanopowders which can control their grain size and sinterability [2].
Ceramic properties defined not only various powder-obtaining conditions but also forming and sintering methods. Last year’s several trends are in the modern production of ceramic materials: developing new methods of nanopowder synthesis; nanoparticle surface modification; and activation of sintering by dopants [1, 3–5].
The initial stage of sintering of various ceramic powders has been investigated by many researchers [1–3, 6]. Application of non-isothermal sintering methods at a constant rate of heating (CRH) plays an important role in the sintering of nanopowders. Using this method, we can examine the initial sintering stage in detail. It is important to investigate the consolidation processes exactly, which are connected with peculiar properties of the sintering of nanoparticles with a small grain size and high specific surface area. WS Young and IB Cutler [7] have investigated the initial sintering behavior in Y-TZP under CRH. J Wang and R Raj have estimated the activation energies at the initial sintering stage of Y-TZP and Al2O3/Y-TZP composite by the CRH method [6, 8].
The main goal of this paper is investigation of the influence of different nanopowder-obtaining conditions on sintering kinetics of ceramic nanocomposites based on Y-TZP. It is especially important to clarify the effect of various nanopowder-obtaining conditions on the sintering process. This data will ensure possibility to control the sintering process of Y-TZP ceramics for producing new ceramic nanocomposites.
Methods
Samples Preparation
We used two kinds nanopowders: TZ-3Y (TOSOH, Tokyo, Japan) containing 3 mol % Y2O3 manufactured by hydrolysis method and 3Y-TZP containing 3 mol % Y2O3 (DIPE of the NASU, Ukraine) produced by co-precipitation method.
The preparation technique according to the manufacture’s data consists several stages: ZrOCl2*8H2O, YCl3 → technical hydrolysis process → drying → calcinations → mechanical milling (48 hours) → spray-drying (hot gas) → nanopowder TZ-3Y (TOSOH).
Mechanical effects are used in technologies based on conventional thermal solid-phase synthesis, mixing the components and increasing the contact surface between the solid particles. Agglomeration degree depends on nanopowder-obtaining method [9]. Mechanical activation in the mills is often used for reducing the aggregation degree and the activation of the particle surface [9].
The preparation technology of the second powder has the following stages: initial salt dissolution ZrOCl2*8H2O, YCl3 → co-precipitation → drying → calcination → nanopowder ZrO2-3 mol % Y2O3 (3Y-TZP, DIPE) without mechanical milling and spray-drying. All the used chemicals have chemical purity. At first, appropriate amounts of Y2O3 were dissolved in nitric acid; then, the zirconium and yttrium salts are mixed via a propeller stirrer for 30 min and subsequently added to an aqueous solution of the precipitant (25 % NH4OH) with constant stirring. Sediments were mixed for 1 h at room temperature at a pH of 9. The sediments were then repeatedly washed and filtered with distilled water. For chloride salts, washing was carried out until a negative test for C1− ions with a silver nitrate solution. After washing and filtration, the hydrogel was dried in a microwave furnace. The calcination of dried zirconia hydroxides was carried out in resistive furnaces at temperature 1000 °C for 2 h.
The powders obtained after calcination were characterized by X-ray diffraction (XRD) employing a DRON-3 diffractometer with Cu-K-α radiation. Fitting and analysis of the XRD curves were made by Powder Cell software for Windows version 2.4. The powders were also studied by transmission electron microscopy (TEM) (JEM 200A, JEOL, and Japan), and the observed average particle size was compared with the value obtained by XRD. Reliable data were obtained by analyzing data from 30 TEM fields for both powders.
Further, all the powders were pressed uniaxially into cylinder specimens with the following dimensions: diameter 6 mm, height 15–17 mm. After that, the powder compacts were treated by a high hydrostatic pressure of 300 MPa.
The shrinkage data of the sintering powder compacts was obtained using a dilatometer (NETZSCH DIL 402 PC). The dilatometer was calibrated using a standard sample of Al2O3. Measurements of shrinkage by the CRH method were carried out in the range from room temperature to 1500 °C with different heating rates of 2.5, 5, 10, 20 °C/min. Upon reaching the temperature of 1500 °C, the samples were cooled at a constant rate. Thermal expansion of each sample was corrected with the cooling curve by the method described in [9]. It was confirmed that the shrinkage proceeded isotropically. The density of the sintered samples was measured using the Archimedes method.
The Analytical Method for the Determination of the Sintering Mechanism at the Initial Sintering Stage
The diffusion mechanism and activation energy of diffusion at the initial sintering stage were determined by the same analytical method as that in the papers [1, 2, 4, 5]. The initial sintering stage is not more than 4 % of relative shrinkage. In this temperature ranges, interparticle contacts begin to form only and grain growth is negligible. Assuming isotropic shrinkage of the samples, the density ρ (T) at a given temperature T is given by the following equation [6]:
where L f is the final length; L(T), the length of the sample at temperature T; and ρf, the final density of the sample measured by hydrostatic method. For investigations, the relative shrinkage no greater than 4 % was selected.
The diffusion mechanism and activation energies were determined by the analytical method described in [4]. Sintering rate at the initial sintering stage is expressed by the equation:
ΔL = (L 0−L) is the change in length of the specimen; K, the numerical constant; Ω, the atomic volume; D, the diffusion coefficient; γ, the surface energy; t, the time; T, the temperature; k, the Boltzmann constant; a, the particle radius; and parameters n and p, the order depending on diffusion mechanism. Since value \( {\left(\frac{\varDelta L}{L_0}\right)}^{\frac{1}{n}} \) may be expressed as a density function F′ (ρ), we obtain:
The equation for the sintering rate at the initial stage in the CRH process can be separated into temperature-dependent, grain size-dependent, and density-dependent quantities as follows (J Wang and R Raj) [6, 8, 10] :
The following sintering rate equation that is applicable to CRH data is derived from Eq. (2)
Here, Q is the activation energy; R is the gas constant; f (ρ, n) is the density function that depends on n; c = dT/dt is the heating rate; and D 0 is the pre-exponential term defined as D = D 0exp(−Q/RT).
Equation (4) corresponds to the sintering rate equation derived by J Wang and R Raj. Using the slope S 1 of the Arrhenius-type plot of ln[T(dT/dt)(dρ/dT)] against 1/T at the same density, the Q is expressed as
From Equation (4), Yang and Cutler derived the following sintering rate equation [7]:
Using the slope S 2 of the Arrhenius-type plot of ln[T 2 − n d(ΔL/L 0)/dT] against 1/T, we found:
From Equations (7) and (9), we found the order of diffusion mechanism:
Here, n is in the range of 0.31–0.50. According to two-sphere shrinkage models proposed by several researchers [2, 10], the n value ranges of grain boundary diffusion (GBD) and volume diffusion (VD) are 0.31–0.33 and 0.40–0.50, respectively.
Results and Discussion
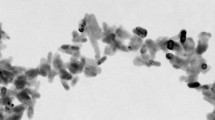
In Fig. 1, we can see the TEM images of nanopowder structure: (a) 3Y-TZP (DIPE) and (b) TZ-3Y (TOSOH). The initial characteristics of nanopowders TZ-3Y (TOSOH) and 3Y-TZP (DIPE) are given in Table 1. As it can be seen, after calcinations of 3Y-TZP (DIPE), we obtained the powder with practically equal particle sizes 31 ± 1.5 nm for 3Y-TZP (DIPE) and 27 ± 1.4 nm for TZ-3Y (TOSOH). The agglomeration degree of the 3Y-TZP (DIPE) nanopowder was larger in comparison with that of TZ-3Y (TOSOH). This means that the powders have different sinterability, what may be due to the different surface states of nanopowders obtained under different conditions. It should be noted that agglomerates in the 3Y-TZP (DIPE) nanopowder are soft and easily destroyed under mechanic and high hydrostatic pressure effects [9]. In comparison with the 3Y-TZP (DIPE) powder, TZ-3Y (TOSOH) was processed by mechanical milling, i.e., their surface was strongly activated. As shown in Fig. 2, the amounts of monoclinic phase in the nanopowder TZ-3Y (TOSOH) are slightly larger than those in 3Y-TZP (DIPE). It is also due to the milling of powder TZ-3Y (TOSOH).

Transmission electron microscopy (TEM) images of nanopowder structures. a 3Y-TZP (DIPE). b TZ-3Y (TOSOH)

X-ray diffraction pattern of a 3Y-TZP (DIPE) and b TZ-3Y (TOSOH)
Figure 3 shows the temperature dependence of the relative shrinkage (dL/L 0) and shrinkage rate (ΔL/dt) of the samples 3Y-TZP (DIPE) and TZ-3Y(TOSOH) at a heating rate of 10 °C/min from room temperature to 1500 °C. As it can be seen, the onset sintering temperatures of shrinkage of these samples are different. In samples TZ-3Y (TOSOH), shrinkage begins earlier at 1010 °C, then shrinkage of samples 3Y-TZP (DIPE) begins at 1075 °C. The highest rate of shrinkage was achieved at temperatures of 1217 and 1256 °C in samples 3Y-TZP and TZ-3Y (TOSOH), respectively. It can be seen that the shrinkage rate of sample 3Z-3Y (DIPE) is more intensive than that of sample TZ-3Y (TOSOH).

Temperature dependence of shrinkage and the shrinkage rate for the samples 3Y-TZP (DIPE) and TZ-3Y (TOSOH) at heating rate 10 °C/min
Figure 4a, b shows the density change of the samples with increasing the sintering temperature from 1300 to 1800 K. In Fig. 5, we can see the temperature dependence of the densification rate (dρ/dT) for the samples (a) TZ-3Y (TOSOH) and (b) 3Y-TZP (DIPE) at different heating rates of 2.5, 5, 10, and 20 °C/min. For samples (a) TOSOH TZ-3Y and (b) 3Y-TZP (DIPE), densification rate curves have shifted to higher temperatures with increasing heating temperature. Densification rate of the samples is different at the same sintering temperatures. In sample TZ-3Y (TOSOH), densification rate is much higher than that in sample 3Y-TZP (DIPE).

Temperature dependence of density of powders a 3Y-TZP (DIPE) and b TZ-3Y (TOSOH) at different heating rates

Temperature dependence of densification rate of samples a 3Y-TZP (DIPE) and b TZ-3Y (TOSOH) at different heating rates
The diffusion mechanism of sintering samples TZ-3Y (TOSOH) and 3Y-TZP (DIPE) was determined by the method described in articles [1, 4, 5, 10] using equations (4, 5, 6, 7, 8, and 9) and Arrhenius-type plots presented in Fig. 6. The obtained activation energy and the order of diffusion mechanisms (n = 0.33–0.51) for the samples are shown in Table 2.

Arrhenius-type plots of samples a 3Y-TZP (DIPE) and b TZ-3Y (TOSOH)
As shown in Table 2 we found that the sample 3Y-TZP (DIPE) was sintered according to the VD mechanism (n = 1/2). And the sample TZ-3Y (TOSOH) was sintered by the grain boundary diffusion (GBD) mechanism (n = 1/3). We can conclude that in samples 3Y-TZP (DIPE), mass transfer process is more intensive than in samples TZ-3Y (TOSOH), which was confirmed by higher sintering rate at the initial stage of sintering.
Conclusions
In the present study, the authors investigated the kinetics of the initial sintering stage of Y-TZP nanopowders manufactured by the hydrolysis method with final processing in mill and co-precipitation method. We used the analytical method applicable to the experimental data obtained at the constant rate of heating (CRH) and found that nanopowder-obtaining conditions significantly affect mass transfer mechanisms at sintering, as well as the activation energy of sintering and the densification rate of zirconia nanopowders. In case of the 3Y-TZP (DIPE) nanopowder obtained by co-precipitation, the sintering occurs predominantly according to the volume diffusion VD mechanism, and in case of the TZ-3Y (TOSOH) nanopowder prepared by the method of technical hydrolysis, the sintering occurs predominantly according to the grain boundary diffusion GBD mechanism.
Since the technical hydrolysis and co-precipitation process are similar and sample characteristics, such as surface area, particle size, and phase structure of nanopowders, are nearly the same, however, predominant diffusion mechanisms of sintering at the initial stage appeared different. Strong surface activation predetermined implementation of grain boundary mechanism at the initial stage of sintering, which requires higher activation energy. It is known that a volume sintering mechanism is preferred due to the relatively low activation energy and the possibility of reducing the sintering temperature of ceramics.
Abbreviations
- 3Y-TZP:
-
3 mol % yttria-stabilized tetragonal zirconia polycrystal
- CRH:
-
constant rate of heating
- DIPE:
-
Donetsk Institute for Physic and Engineering
- GBD:
-
grain boundary diffusion
- NAS:
-
National Academy of the Science
- TEM:
-
transmission electron microscopy
- TZ-3Y (TOSOH):
-
3 mol% yttria-stabilized tetragonal zirconia (Tosoh Corporation Production)
- VD:
-
volume diffusion
- XRD:
-
X-ray diffraction
References
Matsui K (2008) Sintering kinetics at constant rates of heating: mechanism of silica-enhanced sintering of fine zirconia powder. J Am Ceram Soc 91:2534–2539
Johnson DL (1969) New method of obtaining volume, grain-boundary, and surface diffusion coefficients from sintering data. J Appl Phys 40:192–200
Matsu K (2007) Sintering kinetics at constant rates of heating: mechanism of silica-enhanced sintering of fine zirconia powder. J Am Ceram Soc 91:2534–2539
Matsui K, Tanaka K, Enomoto N, Hojo J (2006) Sintering kinetics at constant rates of heating: effect of Al2O3 on the initial sintering stage of yttria-stabilized cubic zirconia powder. J Ceram Soc Jpn 114:763–768
Wang J, Raj R. (1990) Estimate of the Activation Energies for Boundary Diffusion from Rate-Controlled Sintering of Pure Alumina, and Alumina Doped with Zirconia or Titania. J Am Ceram Soc 73:1172.
Wang J, Raj R. J (1991) Activation Energy for the Sintering of Two-Phase Alumina/Zirconia Ceramics. J Am Ceram Soc 74:1959.
Young WS, Cutler IB. (1970) Initial Sintering with Constant Rates of Heating. J Am Ceram Soc 53: 659.
Johnson DL. (1969). New Method of Obtaining Volume, GrainBoundary, and Surface Diffusion Coefficients from Sintering Data. J Appl Phys 40:192.
Lukin E. (1996). Modern high-density oxide ceramics with controlled microstructure. Substantiation the principles of selection of builders that affect the degree of sintering of oxide ceramics. Refractories and technical ceramics, № 5.
Matsui K, Yamakawa T, Uehar M, Enomoto N, Hojo J (2008) Sintering mechanism of fine zirconia powders with alumina added by powder mixing and chemical processes. J Mater Sci 43:2745–275
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing Interests
The authors declare that they have no competing interests.
Authors’ Contributions
ML conceived and designed the experiments, analyzed the data, and wrote the manuscript. ID produced the main idea for writing this manuscript and improved it. TK participated in the data analysis and revised the final edition of the manuscript. GV performed the XRD and TEM experiments. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Lakusta, M., Danilenko, I., Konstantinova, T. et al. Influence of Obtaining Conditions on Kinetics of the Initial Sintering Stage of Zirconia Nanopowders. Nanoscale Res Lett 11, 238 (2016). https://doi.org/10.1186/s11671-016-1452-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s11671-016-1452-3