
Abstract
We have performed a detailed characterization study of electrochemically etched p-type GaAs in a hydrofluoric acid-based electrolyte. The samples were investigated and characterized through cathodoluminescence (CL), X-ray diffraction (XRD), energy-dispersive X-ray spectroscopy (EDX), and X-ray photoelectron spectroscopy (XPS). It was found that after electrochemical etching, the porous layer showed a major decrease in the CL intensity and a change in chemical composition and in the crystalline phase. Contrary to previous reports on p-GaAs porosification, which stated that the formed layer is composed of porous GaAs, we report evidence that the porous layer is in fact mainly constituted of porous As2O3. Finally, a qualitative model is proposed to explain the porous As2O3 layer formation on p-GaAs substrate.
Similar content being viewed by others


Background
GaAs nanostructures are becoming more widespread in many applications including optoelectronic devices [1], solar cells [2], light-emitting diodes (LEDs) [3], tunnel field-effect transistors (TFETs) [4], thermoelectric devices [5], and biosensing [6]. Top-down and bottom-up methods are two approaches used to produce GaAs nanostructures by a variety of physical [7, 8], chemical [9], and electrochemical etching techniques [10]. GaAs nanostructures can be obtained in different forms and shapes including quantum wells [11], nanowires [12], quantum dots (QDs) [13], quantum dot molecules [14], quantum rings (QRs) [15], nanodiscs [16], coupled ring/disks [17], nanopillars [18], nanoholes [19], and nanopores [20]. Electrochemically formed porous GaAs nanostructures are particularly attractive for many applications due to their unique nanoscale properties and high surface-to-volume ratio. Furthermore, the applications of porous GaAs are very broad ranging from antireflective coating for GaAs solar cells [21], virtual substrate for InGaAs due to its weak elastic properties [22], and temporary carrier to reduce the weight of the solar cell by layer transfer processes [23, 24]. It was even shown that porous GaAs are good candidates to obtain fast response to humidity sensing [25]. So far, the porous morphologies obtained from the electrochemical etching of p-doped GaAs are quite different from that of the n-doped type. For n-GaAs, the material pore density, pore dimension, and layer structure depend on the doping density and the crystallographic orientation of the wafer. High aspect ratio triangular pore arrays along the (111) crystallographic direction have been observed by several groups in n-GaAs [26] and [27]. However, for p-GaAs, uniformly distributed mesopores are obtained. The argument is that holes are the majority carriers in p-type substrates and are certainly omnipresent, which will lead to a uniform dissolution even without backside illumination [28]. It is generally believed in the literature that electrochemical etching of p-GaAs leads to porous GaAs formation as it produces nanometric crystallites [29]. However, until now, no detailed analysis of the chemical composition of such layers has been reported. In this work, we report such an analysis by using various characterization techniques to reveal the chemical nature of the porous p-GaAs layer. The spatial and spectral distribution of cathodoluminescence (CL) in bulk and porous samples is presented. Energy-dispersive X-ray spectroscopy (EDS) and X-ray photoelectron spectroscopy (XPS) analysis were performed to confirm the chemical composition. Finally, X-ray diffraction (XRD) was used to determine crystallographic properties of the porous layer.
Methods
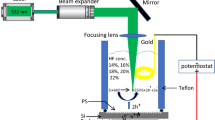
Porous GaAs layers are formed by electrochemical etching on highly doped p-type wafers (Zn-doped, resistivity = 2.10−3 ohm.cm) with (001) crystal orientation supplied by AXT. After immersing the GaAs wafer in an electrolyte consisting of a 49 % HF/water solution, the etching process is activated via direct current. The characteristics of the surfaces are extracted from SEM images using a Zeiss scanning electron microscope, operated from 1 to 20 kV and equipped with a field emission gun. Cross-section images are obtained by cleaving the samples. The bulk porosity is determined through gravimetric measurements, where the sample is weighed by a high precision scale (±0.1 mg) before (m 1) and after (m 2) the etching process. Afterwards, the wafers are introduced for 10 s in H2O:H2O2:H3PO4 (140:2.5:1) in order to selectively remove the porous layer. The remaining mass of the substrate is obtained by weighing the sample again (m 3). The porosity (P) is given by P% = (m 1 − m 2)/(m 1 − m 3). The cathodoluminescence (CL) spectra and images are acquired at room temperature, in the same SEM setup as the one used for imaging the layers. Our CL system, in association with a spectrometer, allows monochromatic CL (GATAN MonoCL2) imaging as well as acquisition of CL spectra on localized spots of a sample with a spectral resolution of 0.5 nm. The accelerating voltage used in the CL characterization is 20 KeV. Surface chemistry is analyzed by XPS Kratos Axis Ultra DLD with monochromatic Al Kα (1486.7 eV) X-ray source and an oval beam size of 300 × 700 μm in diameter. The electron take-off angle is fixed at 60° and the vacuum pressure is below 10−9 Torr during spectra data acquisition. Survey XPS data are acquired over 1200 eV with pass energy of 160 eV and a resolution of 1 eV. High-resolution XPS spectra are obtained at Ga 3d, As 3d, O 1 s, F 1 s, and C 1 s with a pass energy of 20 eV. Binding energies, peak areas, and atom concentration ratios are obtained using CasaXPS. The chemical composition of the exposed surface is verified also by energy-dispersive X-ray (EDX) spectroscopy. Powder X-ray diffractograms have been measured with a Philips X’Pert diffractometer. Samples are prepared simply by crushing porous layer and measured on a standard single crystal.
Results and Discussion
Electrochemical Calibration and Morphology
Calibrations of the porous layer parameters such as porosity and etching rate have been done by varying the current density in the electrochemical process, all other conditions being the same. Figure 1a, b illustrates the dependence of the porosity and the etching rate on the current density. Similarly to porous Si and Ge [30, 31], the etching rate increases linearly with increasing current density. The porosity initially increases rapidly, until it gradually saturates at around 80 %. During the drying process, cracks appear over the entire surface of the sample. The origin of the cracks can be attributed to the high mechanical stresses generated when the water is evaporated [32, 33]. The SEM image of GaAs after porosification is shown in Fig. 2; we can see the presence of micro-crystallites, with sharp edges, dispersed on the surface of the porous layer and having sizes of several hundred nanometers that are attributed to As2O3 precipitates as it was already observed by Smeenk et al. [34]. The resulting clusters show the pyramidal and prismatic morphology with a preferential orientation along the (111) plane corresponding to tiny octahedra As2O3 crystals [35, 36]. High-resolution SEM image of the porous surface shows a randomly and uniformly distributed mesopores structure. The nanocrystallite size is estimated to be in the range of 10–20 nm.

Etching rate (a) and porosity evolution (b) versus the current density of anodically etched p-GaAs in a 49 % HF solution

Planar-view SEM image of anodically etched p-GaAs layer formed in 49 % HF solution at 2 mA/cm2. The inset shows high-resolution SEM image on the porous surface
Luminescence of Porous Layer
Because of the small crystallite size (in the range of 10–20 nm) that composes the porous layer, it is expected that such layer should emit radiation above the GaAs bandgap due to the quantum confinement effect [37]. In order to investigate this effect, we performed cathodoluminescence microanalysis of a GaAs sample before and after the porosification process. CL spectra from the reference sample show luminescence in the near infrared region with a maximum intensity at 860 nm as shown in Fig. 3. The peak is correlated to the interband recombination process of exited charge carriers across the direct bandgap of p-GaAs (1.43 eV) at room temperature [38]. After the porosification process, the CL intensity was considerably attenuated and no detectable shift in the peak was observed despite the low crystallite size of the porous material. This indicates that no quantum confinement effect is created within the crystallites. Which is in contrast with the results obtained by Lockwood et al. who observed luminescence in the infrared (~840 nm) and green (~540 nm) PL peak wavelengths after anodic treatment in HCl solution that are consistent with an assignment to quantum confinement effects in GaAs micro- and nanocrystallites, respectively. The previous observations suggest that the size distribution of the pores correlated qualitatively with the intensities and the positions of the PL peaks, whereas variations in the chemical composition at the surface show no systematic correlation [39, 40]. The drastic decrease in the GaAs peak intensity could be due to different reasons, such as (i) the quenching of the CL due to the surface state, (ii) fractional amorphization of material during anodization, and (iii) change in chemical composition. The later assumption will be verified in the following paragraphs. However, less attention has been paid to the investigation of the direct correlation between luminescence properties and morphology features of the porous GaAs layer. The morphology and CL characteristics of porous layers reported on the spatial mapping of the intensity (Fig. 4) reveal luminescence only around the location of cracks formed during the porosification process. The cross-sectional CL maps also show no luminescence coming from the porous layer. These results suggest that the CL signal is coming from the underlying bulk GaAs material, which is exposed directly in the cracked areas or by the penetration of some electrons through porous layer at high accelerating voltage (20 KeV). The porous layer does not seem to generate detectable luminescence.

Cathodoluminescence spectra at room temperature for the p-GaAs reference substrate and the porous p-GaAs with 20 keV at room temperature formed in 49 % HF solution at 2 mA/cm2

SEM and CL micrographs taken from the top surface (a, b) and cross section (c, d) of porous p-GaAs with 20 keV at room temperature formed in 49 % HF solution at 2 mA/cm2
Chemical Compositions
The absence of size-related luminescence could be due to non-radiative recombinations or to a drastic change in the chemical nature of porous material during anodization. We have investigated this question by verifying that the porous material stoichiometry remained similar to that of GaAs after porosification. Figure 5 shows EDX spectra of the GaAs bulk reference sample and the porous GaAs sample. We notice that the ratio between the Ga- and As-related emissions is changed for the porous layer. It seems that the porous material is enriched with As relative to the reference sample and there is an oxygen-related signal that appears as well. A similar EDX study shows a high decrease of Ga peak on crystalline particles during the anodic dissolution of GaAs in HF solution [41, 42]. An oxygen peak is also present in the porous spectrum indicating a high concentration of oxygen in the film. The EDX mapping of the top of the porous sample confirms the presence of clusters with only As and O contents as shown in Fig. 6. Since the EDX spectrum integrates signals coming from the whole field of view, it is unclear if the oxygen is present in the porous material, or only in the clusters on the surface.

EDX spectra of p-GaAs sample (a) and porous p-GaAs layer formed in 49 % HF solution at 2 mA/cm2 (b)

EDX mapping of a porous p-GaAs layer formed in 49 % HF solution at 2 mA/cm2. a The top view SEM image and the EDX element maps for b As, c Ga, and d O elements
In order to determine the quantitative composition of the porous GaAs layer, we have performed XPS measurements before and after anodization. High-resolution spectra from the As 3d and Ga 3d regions are shown in Fig. 7. A small amount of Ga2O3 and As2O3 is detected on the surface of the reference sample (Fig. 7a, b) due to air exposure [34]. The Ga and As signals appear in the reference sample and are coming from both the base GaAs material as well as the oxides (Ga2O3 and As2O3). The XPS analysis of the porous material (Fig. 7c, d) clearly shows that it consists only of As and arsenic oxide elements. The Ga signal has completely disappeared. However, the Ga presence in EDX spectra is due to the sensitivity of this technique that offers to probe the sample within few microns contrary to XPS measurement with depth analysis of about 8–10 nm. The elemental concentration of the porous layer taken at different surface locations is summarized in Table 1 and indicates that only As and O atoms are present in detectable quantities. This is due to the preferential dissolution of Ga during anodization, leaving a significantly higher As/Ga ratio in the porous layer. This is in agreement with the work of Steer et al. who have shown that Ga dissolved faster than arsenic during polarization in phosphoric acid [41].

Ga 3d and As 3d XPS spectra of p-GaAs reference (a, b) and porous p-GaAs samples (c, d) immediately after anodization in 49 % HF solution at 2 mA/cm2 showing that only the As and O are present in detectable quantities
Electrochemical Mechanisms
In order to explain the behavior of GaAs transformation to porous arsenic oxide under anodic bias, we propose a qualitative model based on the decomposition of compound semiconductors in contact with electrolytes [43]. Figure 8 schematically illustrates the etching mechanism of GaAs in a HF solution. The electrochemical dissolution of semiconductors depends essentially on the symmetry of bi-functional etching agents [44, 45]. The GaAs dissolution in HF is similar to InP in HCl [46]. The first step involves a synchronous exchange of bonds: Ga-F and As-H bonds replace the GaAs surface bonds [47]. The surface becomes H-passivated if the crystal was As-terminated or F-passivated if the crystal was Ga-terminated [48]. This reaction increases the bond polarity of goshawks atoms. Since the surface atoms are doubly bonded to the bulk in the (001) orientations and triply bonded to the bulk in the (111) orientations [49], two and three GaAs bulk bonds must be broken to remove each atom from the lattice following:

Chemical (a, c) and electrochemical (b, d) etching mechanism of GaAs in HF solution
Note that the last GaAs bond should be broken without substitution to produce GaF3 and AsH3.
The second step consists in the formation of arsenic. The elemental arsenic (As) can be obtained by two ways: (i) chemically: from the arsine molecules AsH3 as shown in Fig. 8a. In this case, a non-uniform As layer was obtained due to the difficulty of the nucleation [34] following
(ii) Electrochemically: by involving holes (Fig. 8b) or protons (Fig. 8a) in a redox reaction following a three-hole mechanism proposed for the etching of GaP [50] and InP [46]. For p-GaAs, an accumulation of majority charge carriers (holes) takes place at the surface [51]. In aqueous HF solutions, three holes are required to dissolve one GaAs entity in which only the Ga element is oxidized. This highlights the increasing of the etch rate with the current density applied as shown in Fig. 1. In this case, a uniform As layer was obtained due to the uniform injection of holes following
It was observed that a brown film of As was formed on the electrode during electrochemical etching [34]. The same behavior was revealed in the case of InP etching in HCl solution with a high content of phosphorus but not indium [46]. We suggest that the injection of the holes anodically speeds up this selective etching which explains the low Ga concentration obtained previously in the porous layer.
The last step consists in the formation of As2O3. The presence of water in the electrolyte solution oxidizes elemental As following
Figure 8c shows 3D structures of As2O3 obtained on the electrode surface following reaction (2) or reaction (3) via As transformation to As2O3 by convective diffusion [52]. Additionally, the chemical formation of As2O3 and the porosification process can take place simultaneously, which produces a porous arsenic oxide layer as shown in Fig. 8d. The low crystallite dimension is obtained by the nucleation and the coalescence of the oxide layer formed on the wall pores which will stop the reaction.
Structural Analyses
The last measurement was performed by powder XRD to investigate the crystal structure of the porous film. Figure 9 shows the diffractogram of a crushed porous layer obtained by scraping and grinding the porous sample to ensure that the measurement takes place on the porous layer and not on underlying GaAs substrate. The single-crystal X-ray diffraction measurement is obtained by mounting the crushed porous particles on the goniometer. All other diffraction peaks can be identified as the standard cubic of arsenolite structure (As2O3) with a lattice constant of 11.06 A°. No peak from other expected phases such as GaAs (zincblende) or As4O6 (cubic) are observed within the detection limit of our technique. Effectively, all the peaks correspond to the reflections from (111), (222), (400), and (331) planes of the cubic phase of As2O3 [53]. Hence, the XRD patterns show that the As2O3 film is of a single phase. The intensity of the (111) peak is very strong and its width at half maximum is relatively narrow, indicating a good crystallization state through a large crystallites size.

Powder XRD of porous p-GaAs layer formed in 49 % HF solution at 2 mA/cm2 which corresponds to As2O3 crystal
Conclusions
The nanostructural and chemical nature of porous layers obtained by the anodic etching of p-type GaAs has been investigated. No detectable CL has been attributed to the chemical change of porous GaAs during anodization. Chemical analysis shows that the porous material is depleted of Ga atoms and contains significant amounts of oxygen. Structural data from XRD show that the porous material is composed essentially of crystalline As2O3. An electrochemical process is proposed to explain such a behavior.
Abbreviations
- CL:
-
Cathodoluminescence
- EDX:
-
Energy-dispersive X-ray spectroscopy
- XPS:
-
X-ray photoelectron spectroscopy
- XRD:
-
X-ray diffraction
References
Saxena D, Mokkapati S, Parkinson P, Jiang N, Gao Q, Tan H H, Jagadish (2013) Optically pumped room-temperature GaAs nanowire lasers. Nature Photonics 7: 963–968
Aberg I, Vescovi G, Asoli D, Naseem U, Gilboy JP, Sundvall C, Dahlgren A, Svensson KE, Anttu N, Bjork MT, Samuelson L (2016) A GaAs nanowire array solar cell with 15.3 % efficiency at 1 sun. IEEE J Photovoltaics 6:185–190
Tomioka K, Motohisa J, Hara S, Hiruma K, Fukui T (2010) GaAs/AlGaAs core multishell nanowire-based light-emitting diodes on Si. Nano Lett 10:1639–1644
Ganjipour B, Wallentin J, Borgstrom MT, Samuelson L, Thelander C (2012) Tunnel field-effect transistors based on InP-GaAs heterostructure nanowires. ACS Nano 6:3109–3113
Martin PN, Aksamija Z, Pop E, Ravaioli U (2010) Reduced thermal conductivity in nanoengineered rough Ge and GaAs nanowires. Nano Lett 10:1120–1124
Nazemi E, Aithal S, Hassen WM, Frost EH, Dubowski JJ (2015) GaAs/AlGaAs heterostructure based photonic biosensor for rapid detection of Escherichia coli in phosphate buffered saline solution. Sensors Actuators B Chem 207:556–562
Braive R, Le Gratiet L, Guilet S, Patriarche G, Lemaı̂tre A, Beveratos A, Robert-Philip I, Sagnes I (2009) Inductively coupled plasma etching of GaAs suspended photonic crystal cavities. J Vac Sci Technol B 27:1909–1914
Lee JH, Wang ZM, Abuwaar ZY, Salamo GJ (2009) Design of nanostructure complexes by droplet epitaxy. Cryst Growth Des 9:715–721
Dejarld MT, Shin JC, Chern W, Chanda D, Balasundaram K, Rogers JA, Li X (2011) Formation of high aspect ratio GaAs nanostructures with metal assisted chemical etching. Nano Lett 11:49–54
Ritenour AJ, Levinrad S, Bradley C, Cramer RC, Boettcher SW (2013) Electrochemical nanostructuring of n-GaAs photoelectrodes. ACS Nano 7:6840–6849
Dimastrodonato V, Mereni LO, Young RJ, Pelucchi E (2010) AlGaAs/GaAs/AlGaAs quantum wells as a sensitive tool for the MOVPE reactor environment. J Cryst Growth 312:3057–3062
Ermez S, Jones EJ, Crawford SC, Gradečak S (2015) Self-seeded growth of GaAs nanowires by metal–organic chemical vapor deposition. Cryst Growth Des 15:2768–2774
Heyn C, Stemmann A, Klingbeil M, Strelow C, Koppen T, Mendach S, Hansen W (2011) Mechanism and applications of local droplet etching. J Cryst Growth 323:263–266
Lee JH, Wang ZM, Strom NW, Mazur YI, Salamo GJ (2006) InGaAs quantum dot molecules around self-assembled GaAs nanomound templates. Appl Phys Lett 89:202101
Mano T, Kuroda T, Sanguinetti S, Ochiai T, Tateno T, Kim J, Noda T, Kawabe M, Sakoda K, Kido G, Koguchi N (2005) Self-assembly of concentric quantum double rings. Nano Lett 5:425–42
Kaizu T, Tamura Y, Igarashi M, Hu W, Tsukamoto R, Yamashita I, Samukawa S, Okada Y (2012) Photoluminescence from GaAs nanodisks fabricated by using combination of neutral beam etching and atomic hydrogen-assisted molecular beam epitaxy regrowth. Appl Phys Lett 101:113108
Somaschini C, Bietti S, Sanguinetti S, Koguchi N, Fedorov A (2010) Self-assembled GaAs/AlGaAs coupled quantum ring-disk structures by droplet epitaxy. Nanotechnology 21:125601
Mariani G, Wong PS, Katzenmeyer AM, Léonard F, Shapiro J, Huffaker DL (2011) Patterned radial GaAs nanopillar solar cells. Nano Lett 11:2490–2494
Stemmann A, Heyn C, Köppen T, Kipp T, Hansen W (2008) Local droplet etching of nanoholes and rings on GaAs and AlGaAs surfaces. Appl Phys Lett 93:123108
Föll H, Carstensen J, Langa S, Christophersen M, Tiginyanu IM (2003) Porous III–V compound semiconductors: formation, properties, and comparison to silicon. Phys Status Solidi 197:61–70
Flamand G, Poortmans J (2005) Porous GaAs as a possible antireflective coating and optical diffusor for III-V solar cells. Phys Status Solidi (a) 202:1611–1615
Dimitrakopulos GP, Bazioti C, Grym J, Gladkov P, Hulicius E, Pangrác J, Pacherová O, Komninou P (2014) Misfit dislocation reduction in InGaAs epilayers grown on porous GaAs substrates. Appl Surf Sci 306:89–93
Rojas EG, Terheiden B, Plagwitz H, Hampe C, Tutuc D, Haug R, Brendel R (2009) Mesoporous GaAs double layers for layer transfer processes. Phys Status Solidi A 206:2872–2875
Boucherif A, Beaudin G, Aimez V, Arès R (2013) Mesoporous germanium morphology transformation for lift-off process and substrate re-use. Appl Phys Lett 102:011915
Salehi A, Kalantari DJ, Goshtasbi A (2006) Rapid response of Au/porous-GaAs humidity sensor at room temperature. Conf Optoelectron Microelectron Mater Devices 79:125–128
Grym J, Nohavica D, Gladkov P, Hulicius E, Pangrác J, Piksová K (2013) Epitaxial growth on porous GaAs substrates. Comptes Rendus Chim 16:59–64
Li X, Guo Z, Xiao Y, Um HD, Lee JH (2011) Electrochemically etched pores and wires on smooth and textured GaAs surfaces. Electrochim Acta 56:5071–5079
Tiginyanu I, Langa S, Foell H, Ursachi V (2009) Porous III-V semiconductors, online book: http://www.porous-35.com
Rojas EG (2009) Formation of mesoporous germanium by electrochemical etching for lift-off processes. 24th IEEE Eur. Photovolt. Sol. Energy Conf. Exhib. Hamburg, Germany. doi:10.1109/PVSC.2009.5411208
Blaffart F, Boucherif A, Aimez V, Arès R (2013) Control of mesoporous silicon initiation by cathodic passivation. Electrochem Commun 36:84–87
Bioud YA, Boucherif A, Fafard S, Aimez V, Drouin D, Ares R (2015) Systematic study of mesoporous germanium formation regimes by bipolar electrochemical etching. 9th Int. Conf. Silicon Ep. Heterostruct, Montreal, Canada
Bellet D, Canham LT (1997) Drying of porous silicon, in properties of porous silicon. Inst Eng Technol 5:932
Kowalski SJ, Banaszak J (2013) Modeling and experimental identification of cracks in porous materials during drying. Dry Technol 31:1388–1399
Smeenk NJ, Engel J, Mulder P, Bauhuis GJ, Bissels GMMW, Schermer JJ, Vlieg E, Kelly JJ (2012) Arsenic formation on GaAs during etching in HF solutions: relevance for the epitaxial lift-off process. ECS J Solid State Sci Technol 2:58–65
Ballirano P, Maras A (2002) Refinement of the crystal structure of arsenolite, As2O3. NCS 217:177–178
Anthony JW Arsenolite As2O3 (2005) In Handbook of Mineralogy. Mineral Data Publishing. p 23–24
Kuroda T, Mano T, Ochiai T, Sanguinetti S, Sakoda K, Kido G, Koguchi N (2005) Optical transitions in quantum ring complexes. Phys Rev B 72:1–8
Kittel C (1986) Introduction to solid state physics. Wiley India Pvt. p 139–140
Lockwood DJ, Schmuki P, Labbé HJ, Fraser JW (1999) Optical properties of porous GaAs. Physica E 4:102–110
Schmuki P, Lockwood DJ, Labbé HJ, Fraser JW (1996) Visible photoluminescence from porous GaAs. Appl Phys Lett 69:1620
Steer CA, Weng GS, Luo JL, Ivey DG (2000) Formation of As2O3 during anodic dissolution of GaAs. Electrochem Commun 2:754–761
Bioud YA, Boucherif A, Belarouci A, Paradis E, Drouin D, Ares R (2016) Chemical composition of anodically formed p-type porous GaAs in HF based electrolyte. 10th Int. Conf. Porous Semiconductors - Science and Technology, Tarragona, Spain
Gerische H (1978) Electrolytic decomposition and photodecomposition of compound semiconductors in contact with electrolytes. J Vac Sci Technol 15:1422–1428
Mint W, Gerischer H (1968) The mechanisms of the decomposition of semiconductors by electrochemical oxidation and reduction. Electrochim Acta 13:1329–1341
Wallem-Mattes I, Gerischer H (1969) The Mechanism of the resolution of gallium arsenide by oxidants. Z Phys Chem 64:187–198
Notten PHL (1984) The etching of InP in HCl solutions: a chemical mechanism. J Electrochem Soc 131:2641
Wang W, Lee G, Huang M, Wallace RM, Cho K (2010) First-principles study of GaAs(001)-β2 (2×4) surface oxidation and passivation with H, Cl, S, F, and GaO. J Appl Phys 107:103720
Adachi S, Kikuchi D (2000) Chemical etching characteristics of GaAs(100) surfaces in aqueous HF solutions. J Electrochem Soc 147:4618–4624
Ross FM, Oskam G, Searson PC, Macaulay JM, Liddle JA (1997) Crystallographic aspects of pore formation in gallium arsenide and silicon. Philos Mag A 75:525–539
Schwandt G, Memming R (1968) Electrochemical properties of gallium phosphide in aqueous solutions. Electrochim Acta 13:1299–1310
Schmuki P, Fraser J, Vitus CM, Graham MJ, Isaacs HS (1996) Initiation and formation of porous GaAs. J Electrochem Soc 143:3316–3322
Dai Q, Hu J, Salmeron M (1997) Adsorption of water on NaCl (100) surfaces: role of atomic steps. J Phys Chem B 101:1994–1998
Bozorth RM (1923) The crystal structures of the cubic forms of arsenious and antimonous oxides. J Am Chem Soc 45:1624–1627
Acknowledgements
The authors would like to thank Sonia Blais and Stephane Gutierrez from the Centre de caractérisation des matériaux (CCM) for XPS and EDX characterizations and technical help, SUNLAB research group at the University of Ottawa for scientific discussions, the Natural Sciences and Engineering Research Council of Canada (NSERC), the Regroupement Quebecois sur les Matériaux de Pointe (RQMP), and the Fonds de Recherche du Quebec-Nature et Technologies (FRQNT) for financial support.
Funding
The study was funded by the Natural Sciences and Engineering Research Council of Canada (NSERC), the Regroupement Quebecois sur les Matériaux de Pointe (RQMP), and the Fonds de Recherche du Quebec-Nature et Technologies (FRQNT).
Authors’ contributions
YAB and AB conceived and designed the work. YAB prepared the samples and carried out the measurements. YAB and EP did the CL measurement. DD and RA supervised the research work. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Bioud, Y.A., Boucherif, A., Belarouci, A. et al. Chemical Composition of Nanoporous Layer Formed by Electrochemical Etching of p-Type GaAs. Nanoscale Res Lett 11, 446 (2016). https://doi.org/10.1186/s11671-016-1642-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s11671-016-1642-z