
Abstract
Purpose
The purpose of this study is to develop a simple, effective method to label hepatoma cells with aptamers and then detect them using fluorescent silica nanoparticles (FSNPs).
Method
Streptavidin was conjugated to carboxyl-modified fluorescein isothiocyanate (FITC)-doped silica nanoparticles which were prepared by the reverse microemulsion method. The resulting streptavidin-conjugated fluorescent silica nanoparticles (SA-FSNPs) were mixed with hepatoma cells that had been labeled with biotin-conjugated aptamer TLS11a (Bio-TLS11a). The specificity and sensitivity of the nanoprobes were assessed using flow cytometry and fluorescence microscopy. Their toxicity was assessed in normal human liver cell cultures using the MTT assay, as well as in nude mice using immunohistochemistry.
Results
SA-FSNPs showed uniform size and shape, and fluorescence properties of them was similar to the free FITC dye. SA-FSNPs were able to detect aptamer-labeled hepatoma cells with excellent specificity and good sensitivity, and they emitted strong, photobleach-resistant fluorescent signal. SA-FSNPs showed no significant toxic effects in vitro or in vivo.
Conclusion
The combination of biotin-conjugated aptamers and SA-FSNPs shows promise for sensitive detection of hepatoma cells, and potentially of other tumor cell types as well.
Similar content being viewed by others
Background
Early diagnosis of cancer is key to improving the survival and prognosis of cancer patients [1]. Most cancer detection methods, including blood biochemistry, genetic analysis, and imaging have disadvantages such as low sensitivity, high false-positive rates, high cost or complex procedures [2, 3]. Thus, researchers continue to investigate ways to detect tumor cells simply and effectively in early stages of cancer.
While traditional antibodies against tumor markers can aid in cancer diagnosis, recently developed “chemical antibodies”, which are short sequences of single-stranded DNA or RNA known as aptamers, may prove to be superior. Aptamers specifically recognize targets such as small molecules, protein, virus, bacteria, and whole cells [4, 5]. Aptamers can show higher selectivity and affinity, as well as lower immunogenicity, than traditional antibodies; aptamers are also easier to synthesize, and they can penetrate tissue more rapidly with fewer toxic effects [5–7]. Hundreds of aptamers against tumor cells, most of them labeled with organic dyes, have been described for tumor cell detection [8–13]. One disadvantage of using these fluorescent dye labeled aptamers on their own is that they are rapidly photobleached, severely hindering their clinical usefulness [14].
Recently, the functionalized silica nanoparticles for biosensing have attracted the interest of many researchers [15–18]. And one way to reduce photobleaching of fluorescent-dye labeled aptamers is to conjugate aptamers to the surface of fluorescent silica nanoparticles (FSNPs) [19–21]. With their unique core-shell structure, FSNPs show good biocompatibility, chemical stability, and photostability [22]. Many aptamer-functionalized FSNPs have been reported that they detect tumor cells and show clinical potential for cancer diagnosis [23–25]. However, linking aptamers directly to the nanoparticle surface may destabilize the nanoparticles by making them so large that they are cleared from the circulation [26]. It may also limit the specificity and selectivity of aptamer targeting because of steric hindrance between the target tumor cells and the nanoparticles, such as when aptamer DNA “lies down” on the nanoparticle surface [27]. This is indeed the case with anti-tumor antibodies, which lose much of their sensitivity and specificity after being conjugated to nanoparticles [28].
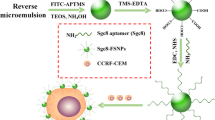
To avoid these potential problems arising from conjugating aptamers directly to FSNPs, we have developed an alternative approach in which the aptamer and FSNP are physically separate but interact via extremely strong biotin-streptavidin interaction (Fig. 1). HepG2 cells are incubated first with biotin-labeled TLS11a aptamer (Bio-TLS11a) and then with streptavidin-conjugated FSNPs (SA-FSNPs). The SA-FSNPs then bind and interact with cells where the biotin-labeled aptamer has bound. This approach avoids the limitations intrinsic to nanoparticle surface modification, and it may allow efficient, sensitive detection of cancer cells in vitro.

Schematic illustration of highly sensitive detection of HepG2 hepatoma cells using a biotin-conjugated aptamer (Bio-TLS11a) and streptadivin-conjugated fluorescent silica nanoparticles (FSNPs)
Methods
Cells and Animals
Human hepatoma cell line HepG2, human normal liver cell line L02, and human embryonic kidney cell line 293T were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). All cell lines were cultured at 37 °C under 5% CO2 in DMEM supplemented with 10% fetal bovine serum (FBS, Hyclone) and penicillin-streptomycin (Gibco, Grand Island, NY, USA).
Female BALB/c nude mice aged 4–6 weeks were obtained from the Guangxi Laboratory Animal Center (Guangxi, China) and housed in laminar flow cabinets under pathogen-free conditions. All experimental protocols were approved by the Animal Ethics Committee of Guangxi Medical University (Nanning, Guangxi, China).
Reagents
Fluorescein isothiocyanate (FITC), cyclohexane, Triton X-100, n-hexanol, bovine serum albumin (BSA), acetone, tetraethyl orthosilicate (TEOS), (3-aminopropyl) triethoxysilane (APTES), 3-aminopropylmethyldimethoxysilane (APTMS), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) N-hydroxysulfosuccinimide sodium salt (sulfo-NHS) and polyoxymethylene were bought from Sigma (St. Louis, MO, USA). Ethanol, dimethyl sulfoxide (DMSO), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), and hematoxylin-eosin (HE) were purchased from Solarbio (Beijing, China). The nuclear dye 4′,6-diamidino-2-phenylindole (DAPI) was purchased from Life Technologies (USA). The biotin-labeled aptamer 5′-bio-(CH2)6-AGTAATGCCCGGTAGTTATTCAAAGATGAGTAGGAAAAGA-3′ (Bio-TLS11a) and FITC-labeled aptamer 5′-FITC-AGTAATGCCCGGTAGTTATTCAAAGATGAGTAGGAAAAGA-3′ (FITC-TLS11a) were synthesized by Shanghai Sangon Biotechnology (Shanghai, China).
Preparation and Characterization of SA-FSNPs
FITC-doped, carboxyl-modified FSNPs were synthesized as described [14, 29, 30]. Briefly, a water-in-oil microemulsion was prepared with FITC, cyclohexane, Triton X-100, n-hexanol, and distilled water, giving rise to FITC-doped silica nanoparticles. These FSNPs were amine-modified using TEOS and APTES; the flocculent precipitate was collected by centrifugation and washed with acetone, followed by deionized water. The precipitate (2 mg) was dissolved in 1 mL of 0.1-M phosphate-buffered saline (PBS, pH 7.4) containing EDC (1 mg) and sulfo-NHS (2.5 mg). When the reaction was complete, 50 μl of streptavidin diluted in PBS was added to the solution, which was incubated at room temperature for 4 h with gentle shaking. The nanoparticles were washed with PBS and then resuspended in 1 ml of 0.05% BSA for 30 min to block free carboxylates, generating SA-FSNPs. The SA-FSNPs were washed three times with PBS and stored at 4 °C. For subsequent experiments, the SA-FSNPs were resuspended in PBS as needed.
The morphology and size distribution of SA-FSNPs were assessed using transmission electron microscopy (TEM; H-7650, Japan). Their photoluminescence was measured using a fluorescence spectrophotometer (FL-7000, Perkin Elmer, USA).
Flow Cytometry of Aptamer-Labeled Cells Mixed with SA-FSNPs
HepG2 or L02 cells (3.0 × 105 cells/ml) were harvested, washed three times with PBS, then incubated with for 30 min either with SA-FSNPs (ca. 0.1 mg, 1 ml) at room temperature or with FITC-TLS11a (100 nM) on ice. In either case, the cells and labeling agents were suspended in binding buffer (200 μl) prepared by supplementing PBS with 4.5 g/L of glucose and 5 mM of MgCl2. Other cell suspensions were incubated with Bio-TLS11a (100 nM) at 4 °C for 30 min, followed by SA-FSNPs (ca. 0.1 mg, 1 ml) at 37 °C for 60 min with gentle shaking. All suspensions were washed three times with PBS, suspended in 500 μl of binding buffer, and then analyzed by flow cytometry (Epics XL, Beckman Coulter, USA) using FLOWJO 7.6 software.
Fluorescence Microscopy of Aptamer-Labeled Cells Mixed with SA-FSNPs
HepG2 and L02 cells were cultured for 12 h in 6-well plates (3 × 105 cells per well). Cells were washed three times with cold PBS, fixed for 15 min with 4% polyoxymethylene, washed with PBS, and then incubated with SA-FSNPs or FITC-TLS11a, or the sequential combination of Bio-TLS11a followed by SA-FSNPs as described above. Finally, cells were stained with DAPI for 90 s, washed with PBS, and analyzed by fluorescence microscopy (DS-Ri1; Nikon Corporation, Tokyo, Japan). Fluorescence intensity was quantitated using Image Pro (Media Cybernetics, Bethesda, MD, USA).
In Vitro Toxicity of SA-FSNPs
Toxicity of SA-FSNPs against 293T or L02 cells was assessed using the MTT assay. Cells (2 × 105 /ml) were cultured overnight in 96-well plates, then treated with SA-FSNPs (0.1, 0.5, or 1.0 mg/ml) for 12, 24, or 48 h. Control cells were treated with PBS. At specific time points, 10 μl of MTT (5 mg/ml) was added to wells, and plates were incubated at room temperature for 4 h in the dark. The medium was discarded, 150 μl of DMSO was added to each well, and plates were incubated for 10 min. Optical density (OD) at 570 nm was measured using an ELISA microplate reader (Thermo Scientific, USA). Cell viability was calculated using the formula:
In Vivo Toxicity of SA-FSNPs
Nude mice received a single tail vein injection of 200-μl PBS or SA-FSNPs (1 mg/ml) (n = 3 animals per group). After 1 week, the animals were sacrificed, and the major tissues (heart, lung, liver, spleen, kidney) were immersed in 10% formaldehyde solution, dehydrated, and paraffin-embedded. Paraffin sections (4 μm thick) were processed using routine methods and stained with HE.
Statistical Analyses
Statistical analysis was performed using Student’s t test and analysis of variance (ANOVA) in GraphPad Prism software (San Diego, CA, USA), with P < 0.05 defined as the significance threshold. Data were shown as mean ± SD or as median (range).
Results and Discussion
Here, we explored the possibility of detecting human hepatoma HepG2 cells, a common cell model for liver cancer studies, using aptamer TLS11a, which was originally selected through the SELEX method to bind specifically to HepG2 cells and which shows promise for targeted diagnostics and therapy of hepatocellular carcinoma [10, 31–33]. In contrast to previous approaches in which the aptamer was conjugated to the surface of FSNPs, potentially limiting the sensitivity of aptamer-based detection, we kept the aptamer and FSNPs physically separate but we conjugated the former to biotin and the latter to streptavidin to allow for strong, specific interaction. Separating aptamer binding to target cells from FSNP binding to aptamer may allow a larger number of aptamers to bind to each target cell, amplifying the fluorescence signal.
Characterization of SA-FSNPs
Transmission electron microscopy showed SA-FSNPs to be nearly monodisperse and spherical, with an average diameter of 75.47 ± 2.52 nm (Fig. 2a). The core-shell structure of silica nanoparticles allows fluorescent dyes such as FITC to be trapped inside [34, 35]. Using rhodamine B in ethanol solution as a reference [36], the fluorescence quantum yields of FITC dye-doped silica nanoparticles were about 0.52. The maximum emission wavelength of free FITC dye and SA-FSNPs was 522 and 525 nm, respectively (Fig. 2b). The emission peak of SA-FSNPs is slightly red-shifted from FITC, which may be due to the loss of energy due to the interaction of silica substrate with the dye [37].

Characterization of SA-FSNPs. a Transmission electron micrograph of SA-FSNPs. b Fluorescence emission spectra of FITC dye and SA-FSNPs
Flow Cytometry of aptamer-Labeled Cells Mixed with SA-FSNPs
To determine whether the synthesized SA-FSNPs can be used as a detection probe for aptamer-labeled cells, HepG2 cells were firstly reacted with Bio-TLS11a. After washing, HepG2 cells was then incubated with SA-FSNPs. L02 cells served as negative cells, and FITC-TLS11a was used as a control probe. For the detection of HepG2 cells, stronger fluorescence intensity was found on Bio-TLS11a combined with SA-FSNPs (Bio-TLS11a + SA-FSNPs) and FITC-TLS11a, while no obvious fluorescence signal was observed on SA-FSNPs alone (Fig. 3a, panel a). Statistical graph of the binding rate of HepG2 cells showed the similar results (Fig. 3a, panel b). Additionally, there was no fluorescence signal on L02 cells after treating with SA-FSNPs alone, FITC-TLS11a and Bio-TLS11a + SA-FSNPs, respectively (Fig. 3b, panel a), in accordance with the results of statistical graph of the binding rate of L02 cells (Fig. 3b, panel b). These results suggest that the sequential combination of Bio-TLS11a with SA-FSNPs can detect HepG2 cells with higher specificity than FITC-TLS11a.

a Flow cytometric detection of HepG2 cells (a) or L02 cells (b) after incubation with SA-FSNPs or FITC-TLS11a or the combination of Bio-TLS11a and SA-FSNPs. b Quantitative analysis of HepG2 cells (a) or L02 cells (b). NS not significant. ** P < 0.01, *** P < 0.001
Fluorescence Microscopy of Aptamer-Labeled Cells Mixed with SA-FSNPs
To allow a more direct visualization of HepG2 detection using our system, we used fluorescence microscopy to examine HepG2 cells incubated with SA-FSNPs or FITC-TLS11a or Bio-TLS11a + SA-FSNPs. As can be seen distinctly in fluorescence images, both FITC-TLS11a and Bio-TLS11a + SA-FSNPs showed green fluorescence on periphery of HepG2 cells, while SA-FSNPs did not. Furthermore, the fluorescence intensity of Bio-TLS11a + SA-FSNPs was stronger than FITC-TLS11a (Fig. 4a). No green fluorescence was observed on L02 cells after incubation with SA-FSNPs alone, FITC-TLS11a and Bio-TLS11a + SA-FSNPs, respectively (Fig. 4b), which was consistent with the analysis of flow cytometry. Therefore, we could agree that aptamer TLS11a could recognize and bind HepG2 cells with high affinity and specificity. Moreover, fluorescence signal from HepG2 cells is owing to the interaction between Bio-TLS11a labeled HepG2 cells and the SA-FSNPs. The SA-FSNPs display stronger fluorescent signals than the FITC-labeled aptamer probably due to the special core-shell structure of silica nanoparticles which allow the fluorescent dyes entrapped inside to prevent them from photodamaging oxidation [38–42].

Fluorescence micrographs of HepG2 and L02 cells after incubation with SA-FSNPs or FITC-TLS11a or the combination of Bio-TLS11a and SA-FSNPs. SA-FSNPs and FITC were examined in the green channel, while DAPI-stained nuclei were examined in the blue channel
Photostability of SA-FSNPs
Fluorescent dye molecules can quench easily after irradiation, limiting their usefulness. Doping fluorophores within porous silica nanoparticles can improve their photostability while maintaining their strong fluorescence emission [34, 35]. We measured the photostability of SA-FSNPs by mixing them with aptamer-labeled HepG2 cells and imaging the cells by fluorescence microscopy after continuous illumination lasting 0, 1, 5, and 10 min. In parallel, cells treated with FITC-aptamer alone were imaged in the same way. Green fluorescence from SA-FSNPs remained clearly visible even after intense irradiation for 10 min, whereas fluorescence from FITC-TLS11a had nearly disappeared after 2 min (Fig. 5). These results are consistent with the idea that fluorescent dye molecules are encapsulated within the silica matrix, where they are kept separate from potential quenchers and photo-oxidizers [39–42].

Photostability of FITC-TLS11a and of the combination of Bio-TLS11a with SA-FSNPs. a Fluorescence micrographs of HepG2 cells labeled with the combination of Bio-TLS11a and SA-FSNPs (upper row) or with FITC-TLS11a alone (lower row) and then continuously irradiated for the indicated periods. b Quantitative analysis of fluorescence intensity after different irradiation periods. *** P < 0.001
Toxicity of SA-FSNPs
We assessed the cytotoxicity of SA-FSNPs on cultures of the normal cell lines 293T and L02. Viability of both cell lines was high according to the MTT assay after incubation with various SA-FSNP concentrations (Fig. 6a), suggesting that SA-FSNP showed minimal cytotoxicity. However, FSNPs have a short half-life in the circulatory system, and the entry of fluorescent dye molecules into the blood may increase the risk of systemic toxicity [43]. Therefore, it is necessary to evaluate the toxicity of SA-FSNPs in vivo. We further studied the in vivo toxicity of SA-FSNPs in nude mice. After intravenous injection of SA-FSNPs for 1 week, tissue sections of the main organs were stained with HE. As shown in Fig. 6b, there were no significant inflammation or necrosis observed on tissue sections. These results confirmed that SA-FSNPs were almost non-toxic to the main organs, showing the potential to be clinically useful as a diagnostic probe.

Toxicity of SA-FSNPs. a 293T and L02 cells in culture were incubated with various concentrations of SA-FSNPs, and cell viability was measured at 12, 24, and 48 h. b Nude mice were treated with PBS or SA-FSNPs, and sections from major organs were stained with hematoxylin-eosin and examined by light microscopy. Magnification, ×400
Conclusions
We have developed an approach to detect hepatoma cells based on a biotin-labeled aptamer and streptavidin-modified FSNPs. The strong affinity and specificity of biotin-TLS11a for HepG2 tumor cells, coupled with the affinity and specificity of biotin for the streptavidin in SA-FSNPs, ensure highly specific and sensitive HepG2 detection. In addition, the fluorescence signal from SA-FSNPs is much stronger and more photostable than the signal from the FITC-labeled aptamer. SA-FSNPs do not show obvious toxic effects in vitro or in nude mice, based on the MTT assay or histology of major organs. This two-step labeling system may be adaptable to the detection of other cancers by changing the aptamer. In addition, this system may become a useful platform for targeted therapy if the nanoparticles can be loaded with anti-tumor drugs or microRNAs.
Abbreviations
- APTES:
-
(3-aminopropyl) triethoxysilane
- APTMS:
-
3-aminopropylmethyldimethoxysilane
- Bio-TLS11a:
-
Biotin-conjugated aptamer TLS11a
- BSA:
-
Bovine serum albumin
- DAPI:
-
4′,6-diamidino-2-phenylindole
- DMSO:
-
Dimethyl sulfoxide
- EDC:
-
1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride
- FITC:
-
Fluorescein isothiocyanate
- FSNPs:
-
Fluorescent silica nanoparticles
- HE:
-
Hematoxylin-eosin
- MTT:
-
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- OD:
-
Optical density
- SA-FSNPs:
-
Streptavidin-conjugated fluorescent silica nanoparticles
- Sulfo-NHS:
-
N-hydroxysulfosuccinimide sodium salt
- TEM:
-
Transmission electron microscopy
- TEOS:
-
Tetraethyl orthosilicate
References
Etzioni R, Urban N, Ramsey S, McIntosh M, Schwartz S, Reid B, Radich J, Anderson G, Hartwell L (2003) The case for early detection. Nat Rev Cancer 3:243–2452
Valente K, Yacoub G, Cappellari JO, Parks G (2016) Metastatic pancreatic acinar cell carcinoma in a younger male with marked AFP production: A potential pitfall on fine needle aspiration biopsy. Diagn Cytopathol doi. doi:10.1002/dc.23610
Sabour L, Sabour M, Ghorbian S (2016) Clinical applications of next-generation sequencing in cancer diagnosis. Pathol Oncol Res doi. doi:10.1007/s12253-016-0124-z
Ganji A, Varasteh A, Sankian M (2016) Aptamer: new arrows to target dendritic cells. J Drug Target 24:1–12
Groff K, Brown J, Clippinger AJ (2015) Modern affinity reagents: recombinant antibodies and aptamers. Biotechnol Adv 33:1787–1798
Meng H, Liu H, Kuai H, Peng R, Mo L, Zhang X (2016) Aptamer-integrated DNA nanostructures for biosensing, bioimaging and cancer therapy. Chem Soc Rev 45:2583–2602
Tan W, Donovan M, Jiang J (2013) Aptamers from cell-based selection for bioanalytical applications. Chem Rev 113:2842–2862
Tang Z, Shangguan D, Wang K, Shi H, Sefah K, Mallikratchy P, Chen HW, Li Y, Tan W (2007) Selection of aptamers for molecular recognition and characterization of cancer cells. Anal Chem 79:4900–4907
Chen H, Medley CD, Sefah K, Shangguan D, Tang Z, Meng L, Smith JE, Tan W (2008) Molecular recognition of small-cell lung cancer cells using aptamers. ChemMedChem 3:991–1001
Shangguan D, Meng L, Cao Z, Xiao Z, Fang X, Li Y, Cardona D, Witek RP, Liu C, Tan W (2008) Identification of liver cancer-specific aptamers using whole live cells. Anal Chem 80:721–728
Li W, Bing T, Wei J, Chen Z, Shangguan D, Fang J (2014) Cell-SELEX-based selection of aptamers that recognize distinct targets on metastatic colorectal cancer cells. Biomaterials 35:6998–7007
Wang Y, Luo Y, Bing T, Chen Z, Lu M, Zhang N, Shangguan D, Gao X (2014) DNA aptamer evolved by cell-SELEX for recognition of prostate cancer. PLoS One 9, e100243
Sefah K, Bae KM, Phillips JA, Siemann DW, Su Z, McClellan S, Vieweg J, Tan W (2013) Cell-based selection provides novel molecular probes for cancer stem cells. Int J Cancer 132:2578–2588
Tan J, Yang N, Hu Z, Su J, Zhong J, Yang Y, Yu Y, Zhu J, Xue D, Huang Y, Lai Z, Huang Y, Lu X, Zhao Y (2016) Aptamer-functionalized fluorescent silica nanoparticles for highly sensitive detection of leukemia cells. Nanoscale Res Lett 11:298
Wu Y, Chen C, Liu S (2009) Enzyme-functionalized silica nanoparticles as sensitive labels in biosensing. Anal Chem 81:1600–1607
Qian J, Zhang C, Cao X, Liu S (2010) Versatile immunosensor using a quantum dot coated silica nanosphere as a label for signal amplification. Anal Chem 82:6422–6429
Yuan L, Xu L, Liu S (2012) Integrated tyramide and polymerization-assisted signal amplification for a highly-sensitive immunoassay. Anal Chem 84:10737–10744
Chen L, Chen C, Li R, Li Y, Liu S (2009) CdTe quantum dot functionalized silica nanosphere labels for ultrasensitive detection of biomarker. Chem Commun (Camb) 21:2670–2672
Wang K, He X, Yang X, Shi H (2013) Functionalized silica nanoparticles: a platform for fluorescence imaging at the cell and small animal levels. Acc Chem Res 46:1367–1376
Smith JE, Medley CD, Tang Z, Shangguan D, Lofton C, Tan W (2007) Aptamer-conjugated nanoparticles for the collection and detection of multiple cancer cells. Anal Chem 79:3075–3082
Herr JK, Smith JE, Medley CD, Shangguan D, Tan W (2006) Aptamer-conjugated nanoparticles for selective collection and detection of cancer cells. Anal Chem 78:2918–2924
Geng J, Liu J, Liang J, Shi H, Liu B (2013) A general approach to prepare conjugated polymer dot embedded silica nanoparticles with a SiO2@CP@SiO2 structure for targeted HER2-positive cellular imaging. Nanoscale 5:8593–8601
Özalp VC, Çam D, Hernandez FJ, Hernandez LI, Schäfer T, Öktem HA (2016) Small molecule detection by lateral flow strips via aptamer-gated silica nanoprobes. Analyst 141:2595–2599
Li H, Mu Y, Lu J, Wei W, Wan Y, Liu S (2014) Target-cell-specific fluorescence silica nanoprobes for imaging and theranostics of cancer cells. Anal Chem 86:3602–3609
Hu H, Dai A, Sun J, Li X, Gao F, Wu L, Fang Y, Yang H, An L, Wu H, Yang S (2013) Aptamer-conjugated Mn3O4@SiO2 core-shell nanoprobes for targeted magnetic resonance imaging. Nanoscale 5:10447–10454
Wu C, Liu J, Zhang P, Li J, Ji H, Yang X, Wang K (2015) A recognition-before-labeling strategy for sensitive detection of lung cancer cells with a quantum dot-aptamer complex. Analyst 140:6100–6107
Algar WR, Krull UJ (2006) Adsorption and hybridization of oligonucleotides on mercaptoacetic acid-capped CdSe/ZnS quantum dots and quantum dot-oligonucleotide conjugates. Langmuir 22:11346–11352
Pathak S, Davidson MC, Silva GA (2007) Characterization of the functional binding properties of antibody conjugated quantum dots. Nano Lett 7:1839–1845
Wang Q, Kang Y (2016) Bioprobes based on aptamer and silica fluorescent nanoparticles for bacteria salmonella typhimurium detection. Nanoscale Res Lett 11:150
Bagwe RP, Yang C, Hilliard LR, Tan W (2004) Optimization of dye-doped silica nanoparticles prepared using a reverse microemulsion method. Langmuir 20:8336–8342
Sun D, Lu J, Zhong Y, Yu Y, Wang Y, Zhang B, Chen Z (2016) Sensitive electrochemical aptamer cytosensor for highly specific detection of cancer cells based on the hybrid nanoelectrocatalysts and enzyme for signal amplification. Biosens Bioelectron 75:301–307
Chen X, Pan Y, Liu H, Bai X, Wang N, Zhang B (2016) Label-free detection of liver cancer cells by aptamer-based microcantilever biosensor. Biosens Bioelectron 79:353–358
Qu L, Xu J, Tan X, Liu Z, Xu L, Peng R (2014) Dual-aptamer modification generates a unique interface for highly sensitive and specific electrochemical detection of tumor cells. ACS Appl Mater Interfaces 6:7309–7315
Lee JE, Lee N, Kim H, Kim J, Choi SH, Kim JH, Kim T, Song IC, Park SP, Moon WK, Hyeon T (2010) Uniform mesoporous dye-doped silica nanoparticles decorated with multiple magnetite nanocrystals for simultaneous enhanced magnetic resonance imaging, fluorescence imaging, and drug delivery. J Am Chem Soc 132:552–557
Lee JE, Lee N, Kim T, Kim J, Hyeon T (2011) Multifunctional mesoporous silica nanocomposite nanoparticles for theranostic applications. Acc Chem Res 44:893–902
Karstens T, Kobs K (2002) Rhodamine B and rhodamine 101 as reference substances for fluorescence quantum yield measurements. Journal of Physical Chemistry 84:1871–1872
Taton TA, Mirkin CA, Letsinger RL (2000) Scanometric DNA array detection with nanoparticle probes. Science 289:1757–1760
Cai L, Chen Z, Chen M, Tang H, Pang D (2013) MUC-1 aptamer conjugated dye-doped silica nanoparticles for MCF-7 cells detection. Biomaterials 34:371–381
Liu A, Wu L, He Z, Zhou J (2011) Development of highly fluorescent silica nanoparticles chemically doped with organic dye for sensitive DNA microarray detection. Anal Bioanal Chem 401:2003–2011
Burns A, Sengupta P, Zedayko T, Baird B, Wiesner U (2006) Core/shell fluorescent silica nanoparticles for chemical sensing: towards single-particle laboratories. Small 2:723–726
Zhao X, Bagwe RP, Tan W (2004) Development of organic-dye-doped silica nanoparticles in a reverse microemulsion. Adv Mater 16:173–176
Liu F, Ni ASY, Lim Y, Mohanram H, Bhattacharjya S, Xing B (2012) Lipopolysaccharide neutralizing peptide-porphyrin conjugates for effective photoinactivation and intracellular imaging of gram-negative bacteria strains. Bioconjugate Chem 23:1639–1647
Jo H, Her J, Ban C (2015) Dual aptamer-functionalized silica nanoparticles for the highly sensitive detection of breast cancer. Biosens Bioelectron 71:129–136
Acknowledgements
The authors acknowledge support from the Key Project of National Natural Science Foundation of China (No. 81430055), the Program for Changjiang Scholars and Innovative Research Teams at University (Ministry of Education of China, IRT_15R13), the International Cooperation Project of the Ministry of Science and Technology of China (20 15DFA31320), and the Project for Innovative Research Teams (Guangxi Natural Science Foundation, 2015GXNSFFA139001).
Authors’ Contributions
ZXH, YXZ, and XLL designed the experiments. ZXH, ZQL, and YWW prepared the nanoparticles. RZ, JHZ, XXL, and NY performed the experiments. JPL, WY, and YH analyzed the data. ZXH and JTT wrote the manuscript. All authors read and approved the final manuscript.
Competing Interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Hu, Z., Tan, J., Lai, Z. et al. Aptamer Combined with Fluorescent Silica Nanoparticles for Detection of Hepatoma Cells. Nanoscale Res Lett 12, 96 (2017). https://doi.org/10.1186/s11671-017-1890-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s11671-017-1890-6