
Abstract
Half-metallicity rising from the s/p electrons has been one of the hot topics in spintronics. Based on the first-principles of calculation, we explore the magnetic properties of the B-doped graphitic heptazine carbon nitride (gh-C3N4) system. Ferromagnetism is observed in the B-doped gh-C3N4 system. Interestingly, its ground state phase (BC1@gh-C3N4) presents a strong half-metal property. Furthermore, the half-metallicity in BC1@gh-C3N4 can sustain up to 5% compressive strain and 1.5% tensile strain. It will lose its half-metallicity, however, when the doping concentration is below 6.25%. Our results show that such a metal-free half-metallic system has promising spintronic applications.
Similar content being viewed by others

Background
Spintronic devices simultaneously utilize the charge and spin freedom of electrons and have attracted increasing attentions due to their potential use in logic and memory devices [1, 2]. Their performances, however, heavily depends on the spin polarization ratio of currents. There is a pressing need for materials that can generate 100% spin-polarized currents, therefore. Half-metal materials, which can do this at Fermi level E F , are considered as the ideal materials for spintronic devices [3,4,5,6]. Many half-metallic ferromagnets, such as doped manganites [7], double perovskites [8], and Heusler compounds [9, 10], have attracted extensive attentions in recent years. However, these half-metallic materials usually contain transition-metal (TM) and have strong spin-orbit coupling strengths, which result in short spin relaxation times. It is necessary to develop advanced TM-free half-metallic materials with long spin relaxation time, therefore.
Two-dimensional (2D) atomic crystals with planar surfaces have attracted a lot of attentions recently due to their potential application in spintronic devices [11,12,13,14,15,16,17,18,19,20,21,22,23,24]. Graphene and its several 2D analogues, such as hexagonal boron nitride and carbon nitride, have great potential for spintronics because of their exceptional properties, e.g., low dimensionality and electron confinement. Although most of these materials are non-magnetic in nature, there are many ways, such as doping and strain to reach the half-metallic ferromagnetism. For example, B, Al, and Cu embedded trizaine-based g-C3N4 (gt-C3N4) have been reported to be half-metallic [14]. The graphene-like carbon nitride also presents half-metallicity under tensile strain [17]. In addition, the heptazine-based g-C3N4 (gh-C3N4) has received a lot of attentions [25,26,27,28,29,30,31,32,33].
A large number of research works have investigated the electronic and magnetic properties of transition metal incorporated gh-C3N4 systems [11, 28, 30]. These transition metal embedded gh-C3N4 materials have been synthesized at elevated temperature [34,35,36,37,38,39]. Theoretical works show that the transition metals can bind more strongly with gh-C3N4 than with graphene and these systems are metallic [30]. Indrani et al. have systematically investigated the magnetic properties of C-dope gh-C3N4 systems by density functional theory (DFT) calculations [40]. They found that all of these C-dope gh-C3N4 systems are ferromagnetism, and a high energy phase shows strong half-metallicity and 400 K Curie temperature. Recently, Gao et al. [41] have experimentally demonstrated the capacity to fabricate the B-doped gh-C3N4 nanosheets, which present high-temperature ferromagnetism and half-metallicity. Despite of these early works, a systematic theoretical investigation of the B-doped gh-C3N4 is missing. Some fundamental issues such as the effects of doping position and B concentration on the electronic and magnetic properties of gh-C3N4 await clarification. Moreover, the effects of strain also need investigation.
In this work, we systematically investigate the effects of doping positions, B concentrations, and strain on the electronic and magnetic properties of the B-doped gh-C3N4 system through first-principles calculations. The results show that strong half-metallicity can be found in the ground state of B-doped gh-C3N4 (BC1@gh-C3N4). Not only doping positions but also doping concentrations play important roles in inducing half-metallicity. Moreover, the half-metallicity in BC1@gh-C3N4 can sustain up to 5% compressive strain and 1.5% tensile strain. The B-doped gh-C3N4 systems are promising for spintronics, therefore.
Computation Methods
A tetragonal 28 a.u. cell containing two primitive cells of gh-C3N4 as shown in Fig. 1 has been employed to simulate the B-doped gh-C3N4 system. The geometry structure relaxation and static electronic structure calculation are performed by using the VASP package [42, 43], which is based on the density functional theory (DFT). The generalized-gradient approximation (GGA) of the Perdew–Burke–Ernzerhof (PBE) [44] and projector augmented wave (PAW) potentials are used. The cutoff energy is set at 500 eV and a 1 × 9 × 15 Monkhorst-Pack k-points grid is chosen to achieve a balance between the calculation time and the accuracy. All of the geometry structures are fully relaxed. The convergence threshold is set at 10−6 eV in electronic steps and 5 × 10−3 eV/Å in force. In order to avoid the interaction between two adjacent periodic images, the vacuum region along the x-direction is set at 15 Å. To investigate the effects of doping concentrations, a tetragonal 112-atomic supercell composed of 2 × 2 × 1 tetragonal unit cells and a 1 × 5 × 9 Monkhorst-Pack k-points grid are adopted.

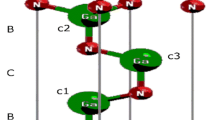
a Schematic representation of pristine gh-C3N4. There are two inequivalent C atoms (C1 and C2) and three inequivalent N atoms (N1, N2, and N3). b The tetragonal 28 a.u. cell of gh-C3N4 is used here to simulate the B-doped gh-C3N4 system (corresponding to 8.33% doping concentration). The black dashed circles indicate the possible B doping sites. c, d The optimized structures of BC1@gh-C3N4 and BC2@gh-C3N4, respectively. Distributions of charge density of spin-up state minus spin-down state for BC1@gh-C3N4 and BC2@gh-C3N4 are also shown here. The red and blue colors label the spin-up and spin-down charges, respectively
Results and Discussions
In a pure gh-C3N4 system, there are two inequivalent C atoms (C1 and C2) and three inequivalent N atoms (N1, N2, and N3) as shown in Fig. 1a. We find the relaxed lattice parameters (a = b = 7.14 Å) of the pure gh-C3N4 agree well with the previous experimental and theoretical reports [40, 45]. The band structure and the corresponding total density of states (DOSs) of gh-C3N4 are shown in Fig. 2a. To further understand the electronic properties of the gh-C3N4, the charge distributions of the edge bands C 1 , V 1 , and the corresponding local density of states are presented in Fig. 2b, c. It can be clearly seen that the bottom of conduction band C 1 is dominated by the π* states of C1, C2, and N3 atoms, which originate from the p x orbitals. However, the top of valence band V 1 is determined by the non-bonding δ states of N2 atoms and the π states of N3 atoms.

a The electronic band structures and the total density of states of pristine gh-C3N4. b The charge distributions of the edge bands C 1 and V 1 (indexed in a). c The orbital-resolved electron density of states projected onto C1 atom, C2 atom, N2 atom, and N3 atom (indexed in b). The energy at the Fermi level is set to zero
A tetragonal unit cell containing 28 atoms of gh-C3N4 (corresponding to 8.333% doping concentration) is employed to simulate the B-doped gh-C3N4 system as shown in Fig. 1b (the red dashed line). After considering early report [31] that the substitution on the C sites (C1 and C2) is more favorable than on the N sites (N1, N2, and N3), only the configurations of B substituting C have been investigated to explore their magnetic properties. As a result, the two different B-doped gh-C3N4 isomers (BC1@gh-C3N4 and BC2@gh-C3N4) are studied. The fully relaxed structures of BC1@gh-C3N4 and BC2@gh-C3N4 are given in Fig. 1c, d, respectively.
The structural stability depends on the extent of cohesive and the system with negative and large absolute cohesive energy has better stability. The cohesive energies (Ecoh) of BC1@gh-C3N4 and BC2@gh-C3N4 have been calculated by using
where Etot is the total energy of a B-doped gh-C3N4 system and E i is the energy of an isolated atom for element i in the same cell. The M i and M are the number of the ith species and the total number of atoms presented in the B-doped gh-C3N4 system, respectively. We find that the cohesive energies are − 6.107 and − 6.097 eV per atom for BC1@gh-C3N4 and BC2@gh-C3N4, respectively. Thus, the BC1@gh-C3N4 phase is energetically favorable. This conclusion agrees well with the previous work [31]. To further study the relative stability of the two B-doped gh-C3N4 systems, the cohesive energies of 2D C2N and gh-C3N4, which have been synthesized experimentally, are calculated and equal to − 6.813 and − 6.091 eV per atom, respectively. Interestingly, both BC1@gh-C3N4 and BC2@ gh-C3N4 have intermediate cohesive energies between C2N and gh-C3N4. Consequently, they should have intermediate structural and mechanical stability.
In order to determine the thermodynamic feasibility and the relative energy cost of BC1@gh-C3N4 and BC2@gh-C3N4 when compared to their pristine 2D analogues, the formation energies have also been calculated using
where Etot, M i , and M are the same as those for the calculation of cohesive energy. μ i is the chemical potential of the ith species. Here, graphene, rhombohedral boron and gaseous nitrogen are used to determine the chemical potentials μ C , μ B , and μ N , respectively. The calculated formation energies are 0.222 and 0.232 eV per atom for BC1@gh-C3N4 and BC2@gh-C3N4, respectively. As a comparison, the formation energy of gh-C3N4 is 0.293 eV per atom. In addition, the calculated E f values of BC1@gh-C3N4 and BC2@gh-C3N4 are slightly lower than gh-C3N4, indicating these B-doped gh-C3N4 isomers can be fabricated. Indeed, the synthesis of B-doped gh-C3N4 has been reported [41].
In order to find out the magnetic ground states of BC1@gh-C3N4 and BC2@gh-C3N4, we have investigated the non-spin polarized (NSP), ferromagnetic (FM), and antiferromagnetic (AFM) states. The results show that FM state is the ground state for the two B-doped gh-C3N4 systems, and their magnetic moments are both 1.0 μ B per unit cell as shown in Table 1. For further understanding of the magnetism of the two B-doped gh-C3N4 systems, the spin-dependent charge densities of BC1@gh-C3N4 and BC2@gh-C3N4 have been investigated and depicted in Fig. 1c, d, respectively. Slightly different from the C-doped gh-C3N4 systems in which the spin density is mainly located at the doped C-sites [40], the spin density of B-doped gh-C3N4 is mainly localized at the 2-fold coordinated N2 atoms, especially the N2 atoms adjacent to the dopant B atoms, as shown in Fig. 1c, d. Because B dopant has one less electron than the substituted C atom, a π defect is induced in B-doped gh-C3N4 system, resulting in 1.0 μ B magnetic moment.
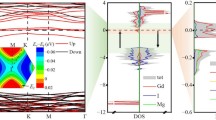
To understand the effects of B doping on the gh-C3N4 systems, we performed the spin polarized band structure and density of states calculations for BC1@gh-C3N4 and BC2@gh-C3N4, as shown in Fig. 3a, d, respectively. The results show that the asymmetry between spin-up and spin-down densities in BC1@gh-C3N4 and BC2@gh-C3N4 induces an obvious magnetism. Interestingly, as shown in Fig. 3a, we find that the BC1@gh-C3N4 systems have a half-metallic property as one of the spin-channels is metallic, whereas the other one is insulating. The band structure and total density of states plots show that the spin splitting occurs close to the Fermi level and two spin-down bands are crossing the Fermi level, while the spin-up ones have a band gap of 1.23 eV. This is mainly because of the large voids present in the gh-C3N4 framework, which lead to the localization of electronic states. The band gap in the spin-up channel of BC1@gh-C3N4 is far larger than the gaps (in one of spin channel) of doped manganites [7], double perovskites [8], Heusler compounds [9, 10], and graphene nanoribbon [46]. The half-metallic strength of the BC1@gh-C3N4 systems can be comparable with the C-doped gh-C3N4 [40]. Such a strong half-metallic system is very promising because the spin-flip transition of carriers from the thermal excitation is not possible. To further explore the origins of the half-metallicity in BC1@gh-C3N4, the charge distributions of the two spin-down bands that across the Fermi level are presented in Fig. 3b. We clearly see that the half-metallicity of BC1@gh-C3N4 mainly comes from the non-bonding δ states of N2 atoms. The local density of states (see Fig. 3c) also shows that the half-metallicity of BC1@gh-C3N4 mainly originates from the p z orbits of N2 atoms along with a partial contribution from the p z orbits of B and N1 atoms. They are in good agreement with the earlier reports on gt-C4N3 [2], where the N orbitals provide a major contribution to the half-metallicity. For the BC2@gh-C3N4, the band structure and total density of states plots (Fig. 3a) also show that spin splitting occurs close to the Fermi level. The spin majority state has a band gap of 1.36 eV. However, the spin minority state shows a 0.016 eV band gap. The charge distributions of the edge bands and local density of states for BC2@gh-C3N4 show that both the valence band edges and the conduction band edges of BC2@gh-C3N4 are dominated by the non-bonding δ states, originating mainly from the p y and p z orbitals of N2 atoms. This means that the non-bonding δ states of N2 atoms are split when a B atom substitutes a C atom in gh-C3N4 system and determine its electronic properties.

a The spin-dependent band structure and the total density of states of BC1@gh-C3N4. b The charge densities of the two bands crossing the Fermi level. c The orbital-resolved electron density of states projected onto B atom, N1 atom, and N2 atom (indexed in b) for BC1@gh-C3N4. d–f are the same with a–c but for BC2@gh-C3N4. The energy at the Fermi level is set to zero
In order to clarify the dependence of half-metallicity in the BC1@gh-C3N4 systems on doping concentrations, a tetragonal 112-atomic supercell of 2 × 2 × 1 tetragonal unit cell has been employed and three different B-doping concentrations (2.083, 4.167, and 6.25%) are investigated, as shown in Fig. 4a, b. As we can see from Fig. 4b, BC1@gh-C3N4 can still sustain the half-metallicity for 6.25% doping concentration. However, it loses its half-metallicity as the doping concentration equal to or low than 4.167%.

a Schematic representations of the tetragonal 112-atomic supercell used to simulate different doping concentrations of BC1@gh-C3N4. b The spin-dependent total density of states of BC1@gh-C3N4 with different doping concentrations. The energy at the Fermi level is set to zero
Strain technology is commonly used to tune the spin properties of a magnetic material, and the strain effect on half-metallicity of a material should be studied. Here, we carried out the density of state calculations for the BC1@gh-C3N4 system under the in-plain biaxial strain. It is found that the half-metallicity strength gradually decreases as the biaxial tensile strain increases. It loses half-metallicity when the biaxial tensile strain reaches 1.5% as shown in the panel of Fig. 5. However, it sustains half-metallicity up to 5% of the biaxial compressive strain (see the right panel of Fig. 5). Thus, this system behaves well under external strain.

The spin-dependent total density of states of BC1@gh-C3N4 (with 8.33% doping concentration) under in-plain biaxial tensile strain (left) and biaxial compressive strain (right), respectively. The energy at the Fermi level is set to zero
Conclusion
Based on density functional theory calculations, the B-doped gh-C3N4 systems have been investigated for potential applications in spintronic devices. Ferromagnetism is observed in all B-doped gh-C3N4 systems. Moreover, a strong half-metallicity is achieved only in the ground state phase, i.e., BC1@gh-C3N4, which results from a spin split of the non-bonding δ states of highly unsaturated 2-fold coordinated N2 atoms. The half-metallicity is lost for low B-doping concentrations. Thus, both selective doping and its concentration play an important role in inducing magnetism and half-metallicity. The half-metallicity in BC1@gh-C3N4 can sustain up to 5% compressive strain and 1.5% tensile strain. These results show that the B-doped gh-C3N4 systems could be a ferromagnetic half-metallic material for magnetic memory and spintronic devices.
References
Wolf SA, Awschalom DD, Buhrman RA, Daughton JM, von Molnár S, Roukes ML, Chtchelkanova AY, Treger DM (2001) Spintronics: a spin-based electronics vision for the future. Science 294:1488–1495
Du A, Sanvito S, Smith SC (2012) First-principles prediction of metal-free magnetism and intrinsic half-metallicity in graphitic carbon nitride. Phys Rev Lett 108:197207
Felser C, Fecher GH, Balke B (2007) Spintronics: a challenge for materials science and solid-state chemistry. Angew Chem Int Ed 46:668–699
Si C, Zhou J, Sun Z (2015) Half-metallic ferromagnetism and surface functionalization-induced metal–insulator transition in graphene-like two-dimensional Cr2C crystals. ACS Appl Mater Interfaces 7:17510–17515
Hod O, Barone VN, Peralta JE, Scuseria GE (2007) Enhanced half-metallicity in edge-oxidized zigzag graphene nanoribbons. Nano Lett 7:2295–2299
Meng B, Xiao W-z, Wang L-l, Yue L, Zhang S, Zhang H-y (2015) Half-metallic and magnetic properties in nonmagnetic element embedded graphitic carbon nitride sheets. Phys Chem Chem Phys 17:22136–22143
Zhao G, Keller H, Prellier W, Kang DJ (2000) Bulk experimental evidence of half-metallic ferromagnetism in doped manganites. Phys Rev B 63:172411
Liu YP, Fuh HR, Wang YK (2013) Study of the half-metallic materials double perovskites Sr2ZnBO6 (B=Tc, Re, Ru, Os, Co, Pd, and Au) via first-principle calculations. J Magn Magn Mater 341:25–29
Yu HL, Yang GW (2011) Elimination of interface states of Co2MnSi/MgO/Co2MnSi magnetic tunneling junction by inserting an Al atomic layer. Appl Phys Lett 98:011910
Yu HL, Zhang HB, Jiang XF, Zheng Y, Yang GW (2012) Transport and magnetic properties of the Co2MnSi/Al/Co2MnSi trilayer. Appl Phys Lett 100:222407
Ghosh D, Periyasamy G, Pandey B, Pati SK (2014) Computational studies on magnetism and the optical properties of transition metal embedded graphitic carbon nitride sheets. J Mater Chem C 2:7943–7951
Xu K, Li X, Chen P, Zhou D, Wu C, Guo Y, Zhang L, Zhao J, Wu X, Xie Y (2015) Hydrogen dangling bonds induce ferromagnetism in two-dimensional metal-free graphitic-C3N4 nanosheets. Chem Sci 6:283–287
Gao D, Liu Y, Song M, Shi S, Xue D (2015) Manifestation of high-temperature ferromagnetism in fluorinated graphitic carbon nitride nanosheets. J Mater Chem C 3:12230–12235
Gong S, Wan WH, Guan S, Tai B, Liu C, Fu BT, Yang SYA, Yao YG (2017) Tunable half-metallic magnetism in an atom-thin holey two-dimensional C2N monolayer. J Mater Chem C 5:8424–8430
Zhang X, Wang A, Zhao M (2015) Spin-gapless semiconducting graphitic carbon nitrides: a theoretical design from first principles. Carbon 84:1–8
Yang B, Zhou H, Zhang X, Zhao M (2015) Electron spin-polarization and band gap engineering in carbon-modified graphitic carbon nitrides. J Mater Chem C 3:10886–10891
Li H, Hu H, Bao C, Hua J, Zhou H, Liu X, Liu X, Zhao M (2015) Tensile strain induced half-metallicity in graphene-like carbon nitride. Phys Chem Chem Phys 17:6028
Brito WH, da Silva-Araújo J, Chacham H (2015) Magnetic properties of C–N planar structures: d0 ferromagnetism and half-metallicity. Phys Chem Chem Phys 17:31995
Choudhuri I, Kumar S, Mahata A, Rawat KS, Pathak B (2016) Transition-metal embedded carbon nitride monolayers: high-temperature ferromagnetism and half-metallicity. Nano 8:14117
Liu B, Wu LJ, Zhao YQ, Wang LZ, Cai MQ (2016) A first-principles study of magnetic variation via doping vacancy in monolayer VS2. J Magn Magn Mater 420:218–224
Liu Y, Liu P, Sun C, Wang T, Tao K, Gao D (2017) P dopants induced ferromagnetism in g-C3N4 nanosheets: experiments and calculations. Appl Phys Lett 110:222403
Daguo G, Zhou Y, Ma R, Wang F, Liu Q, Wang J (2017) Facile synthesis of N-doped graphene-like carbon nanoflakes as efficient and stable electrocatalysts for the oxygen reduction reaction. Nano-Micro Lett 10:29
Wang A, Wang C, Li F, Wong-Ng W, Lan Y (2017) Recent advances of graphitic carbon nitride-based structures and applications in catalyst, sensing, imaging, and LEDs. Nano-Micro Lett 9:47
Fang Y, Wang X (2017) Metal-free boron-containing heterogeneous catalysts. Angew Chem Int Ed 56:15506–15518
Wang X, Maeda K, Thomas A, Takanabe K, Xin G, Carlesson JM, Domen K, Antonietti M (2009) A metal-free polymeric photocatalyst for hydrogen production from water under visible light. Nat Mater 8:76–80
Luan HX, Zhang CW, Li SS, Zhang RW, Wang PJ (2013) First-principles study on ferromagnetism in W-doped graphene. RSC Adv 3:26261–26265
Kan M, Zhou J, Sun Q, Kawazoe V, Jena P (2013) The intrinsic ferromagnetism in a MnO2 monolayer. J Phys Chem Lett 4:3382–3386
Ghosh D, Periyasamy G, Pati SK (2014) Transition metal embedded two-dimensional C3N4–graphene nanocomposite: a multifunctional material. J Phys Chem C 118:15487–15494
Crook CB, Constantin C, Ahmed T, Zhu J-X, Balatsky AV, Haraldsen JT (2015) Proximity-induced magnetism in transition-metal substituted graphene. Sci Rep 5:12322
Zhang Y, Wang Z, Cao J (2014) Prediction of magnetic anisotropy of 5d transition metal-doped g-C3N4. J Mater Chem C 2:8817–8821
Ding K, Wen L, Huang M, Zhang Y, Luc Y, Chen Z (2016) How does the B,F-monodoping and B/F-codoping affect the photocatalytic water-splitting performance of g-C3N4? Phys Chem Chem Phys 18:19217–19226
Zhu B, Zhang J, Jiang C, Cheng B, Yu J (2017) First principle investigation of halogen-doped monolayer g-C3N4 photocatalyst. Appl Catal B Environ 207:27–34
Masih D, Ma Y, Rohani S (2017) Graphitic C3N4 based noble-metal-free photocatalyst systems: a review. Appl Catal B Environ 206:556–588
Zhang G, Lan ZA, Lin L, Lin S, Wang X (2016) Overall water splitting by Pt/g-C3N4 photocatalysts without using sacrificial agents. Chem Sci 7:3062–3066
Zou X, Silva R, Goswami A, Asefa T (2015) Cu-doped carbon nitride: bio-inspired synthesis of H2-evolving electrocatalysts using graphitic carbon nitride (g-C3N4) as a host material. Appl Surf Sci 357:221–228
Zhang G, Zang S, Wang X (2015) Layered Co(OH)2 deposited polymeric carbon nitrides for photocatalytic water oxidation. ACS Catal 5:941–947
Xu J, Long K-Z, Wang Y, Xue B, Li Y-X (2015) Fast and facile preparation of metal-doped g-C3N4 composites for catalytic synthesis of dimethyl carbonate. Appl Catal A Gen 496:1–8
Gao L-F, Wen T, Xu J-Y, Zhai X-P, Zhao M, Hu G-W, Chen P, Wang Q, Zhang H-L (2016) Iron-doped carbon nitride-type polymers as homogeneous organocatalysts for visible light-driven hydrogen evolution. ACS Appl Mater Interfaces 8:617–624
Nasir Baig RB, Verma S, Varma RS, Nadagouda MN (2016) Magnetic Fe@g-C3N4: a photoactive catalyst for the hydrogenation of alkenes and alkynes. ACS Sustain Chem Eng 4:1661–1664
Choudhuri I, Bhattacharyya G, Kumar S, Pathak B (2016) Metal-free half-metallicity in a high energy phase C-doped gh-C3N4 system: a high Curie temperature planar system. J Mater Chem C 4:11530–11539
Gao D, Liu Y, Liu P, Si M, Xue D (2016) Atomically thin B doped g-C3N4 nanosheets: high-temperature ferromagnetism and calculated half-metallicity. Sci Rep 6:35768
Kresse G, Furthmüller J (1996) Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput Mater Sci 6:15–50
Kresse G, Furthmüller J (1996) Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys Rev B Condens Matter 54:11169
Perdew JP, Burke K, Ernzerhof M (1996) Generalized gradient approximation made simple. Phys Rev Lett 77:3865
Ma X, Lv Y, Xu J, Liu Y, Zhang R, Zhu Y (2012) A strategy of enhancing the photoactivity of g-C3N4 via doping of nonmetal elements: a first-principles study. J Phys Chem C 116:23485–23493
Rudberg E, Sałek P, Luo Y (2007) Nonlocal exchange interaction removes half-metallicity in graphene nanoribbons. Nano Lett 7:2211–2213
Acknowledgements
The authors thank National Natural Science Foundation of China, China Postdoctoral Science Foundation, and the Six Talent Peaks Project of Jiangsu for financial support.
Funding
This work was supported by the National Natural Science Foundation of China (NSFC) (Grant No. 61674022) and the China Postdoctoral Science Foundation (2015M570428).
Author information
Authors and Affiliations
Contributions
HY is the first author. HY and YL designed the calculations. HY carried out the calculations and characterizations. XJ, ZS, JF, and XY helped to prepare and correct the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Yu, H., Jiang, X., Shao, Z. et al. Metal-Free Half-Metallicity in B-Doped gh-C3N4 Systems. Nanoscale Res Lett 13, 57 (2018). https://doi.org/10.1186/s11671-018-2473-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s11671-018-2473-x