
Abstract
Hexagonal phase (β) sodium rare earth tetrafluorides (NaREF4, RE = Y, Gd, Lu, et al.) are considered the ideal matrices for lanthanide (Ln) ions doped upconversion (UC) luminescence materials, because they can provide favorable crystal lattice structures for the doped luminescent Ln ions to make intensive emissions. However, the cubic phase (α) NaREF4 always preferentially forms at low reaction temperature in short time as it is dynamically stable. Therefore, it is hard to obtain small sized β-NaREF4 via the traditional solvothermal method. In this paper, small sized β-NaYF4:Yb,Er nanoparticles were synthesized by a heterogeneous-core-induced method via the solvothermal reaction. The heterogeneous α-NaGdF4/β-NaYF4: Yb, Er core–shell structure was confirmed by the local elemental mapping. The formation mechanism of β-NaYF4:Yb,Er shell on the surface of α-NaGdF4 core was explained in detail. We reasoned that a hetero interface with a lower lattice symmetric structure was produced by cation exchanges between the core and shell, which caused the preferential growth of anisotropic hexagonal phase shell. The existence of this hetero interface has also been proven by observation of Gd3+ UC emission.
Similar content being viewed by others

Background
Lanthanide (Ln)-doped rare earth upconversion (UC) luminescence nanoparticles (NPs) have been intensively studied as potential fluorescent probes in biological applications for years due to their attractive luminescence properties, such as sharp emission peaks, photo stability, absence of background noise, and large tissue penetration [1,2,3,4,5,6,7,8]. As UC fluorescence probes in biological detections and imaging, these Ln3+-doped UCNPs should emit strong UC luminescence under infrared excitation in order to achieve high sensitivity and resolution. In the most of cases, the UCNPs are also required to have both small size (sub-50 nm) and hydrophilic surface in order to meet the post biological functionalization [9,10,11,12,13,14]. Among numerous UC materials, Ln3+ activator-doped rare earth fluorides, especially hexagonal phase NaREF4 (RE = Y, Gd, Lu), are considered ideal candidates for probes because of their efficient UC luminescence. Therefore, large numbers of researches have been focused on the synthesis of water soluble, small-sized hexagonal NaREF4 nanoparticles with strong UC luminescence [15,16,17,18,19].
At present, several methods, such as solvothermal reaction [20,21,22,23], high-temperature thermal decomposition [24,25,26,27], and doping strategy [28,29,30,31], have been developed aiming to prepare hexagonal NaREF4 nanocrystals that can possess all above properties. The solvothermal method in which hydrophilic surfactants have commonly been employed, for example, polyvinylpyrrolidone (PVP) or polyethylenimine (PEI) as chelating agents, has been used to synthesize good-water-soluble NaREF4 nanocrystals with small size [21, 32]. But these nanocrystals usually were cubic phase which exhibits less efficient than hexagonal phase counterparts for UC luminescence. Although through extending reaction time, increasing the concentration of fluorine source, or changing synthesis approaches, hexagonal phase nanocrystals could also be obtained, however, the size of these nanocrystals would correspondingly increase up to 100 nm [33, 34]. The high-temperature thermal decomposition method was developed to successfully synthesize pure hexagonal NaREF4 nanocrystals with small size (even ultra-small size of sub-10 nm) [27, 35, 36]. But this synthesis method has usually employed the environmentally unfriendly mixed trifluoroacetates as precursors. Moreover, it required drastic conditions such as sufficiently high reaction temperatures (over 300 °C), rather narrow temperature window of the decomposition (less than 10 °C), waterless, oxygen-free, and inert gas protection, which was generally difficult to be reproduced. Additionally, the as-prepared nanocrystals were hydrophobic, thus requiring further surface engineering to render them hydrophilic [37, 38]. Besides the above approaches, additional doping with other Ln ions could realize the phase and size control of NaREF4 nanocrystals in a facile way [31]. The problem which had to be faced in doping strategy was that the additional doping ions had to be over a certain amount to trigger the cubic-to-hexagonal phase transition. However, large amount of additional Ln ions probably changed the NaREF4 to other NaRE’F4 host.
In 2014, our group reported a novel heterogeneous core/shell strategy to prepare water soluble small-sized hexagonal NaREF4 nanoparticles [39]. In this strategy, small cubic cores were used to induce the growth of heterogeneous hexagonal shells. Two types of core/shell nanoparticles, α-NaLuF4/β-NaYF4:Yb,Er and α-NaYF4/β-NaLuF4:Yb,Er nanoparticles, were prepared. We presumed that the heterogeneous interface between the core and shell owing to cation exchange was the key factor, which caused the shell to grow into hexagonal phase. In this work, we selected cubic NaGdF4 nanocrystals as cores to induce the growth of hexagonal NaYF4 shells according to the heterogeneous core/shell strategy mentioned above. There are several reasons for NaGdF4 to be chosen as core material. First, Gd3+ possesses a large energy gap between its ground state and excited state, making NaGdF4 to be a good host matrix. Pure NaGdF4 cannot absorb 980-nm photons directly; however, if Yb3+ and Er3+ are codoped in NaGdF4 matrix, nanoparticles can emit ultraviolet (UV) UC luminescence through the energy transfer processes from Er3+ to Gd3+ [40,41,42]. In our experiments, NaGdF4 cores were prefabricated and no Yb3+ and Er3+ were doped. If the UC luminescence of Gd3+ could be observed in core/shell nanoparticles, the formation of heterogeneous interface between core and shell would be confirmed. Second, the ions radii of Gd3+ (0.938 Å) is larger than Y3+ (0.9 Å) [31]. It is easy for Y3+ to substitute Gd3+, forming a heterogeneous interface with low lattice symmetry, in our reasoning, thus triggering the growth of β-NaYF4 shell. Compared using NaLuF4 as core material, it might need shorter reaction time to obtain β-NaYF4 shell by using NaGdF4 cores. In our experiments, the heterogeneous α-NaGdF4/β-NaYF4:Yb,Er core/shell structure was certified by various characterizations.
Methods
Materials
The rare earth chloride, including gadolinium chloride (GdCl3), yttrium chloride (YCl3), ytterbium chloride (YbCl3), and erbium chloride (ErCl3), was obtained from Sandong Yutai Rare Earth Technology Co., Ltd. China (all with purity > 99.9%). Sodium chloride (NaCl, AR), potassium fluoride (KF, AR), and ethylene glycol (EG, AR) were bought from Shanghai Shabo Chemical Technology Co., Ltd. China. Polyvinylpyrrolidone K-30 (PVP, 58,000 g/mol) was obtained from Aldrich. All chemicals were used as received and without further purification.
Synthesis of NaGdF4 Core Nanocrystals
Metal chloride (RECl3, NaCl) stock solutions were prepared by dissolving the corresponding metal chloride in EG. Polyvinylpyrrolidone K-30 (PVP, 0.5 g) was dissolved in EG (10 mL) to form a transparent solution. GdCl3 (1 mmol) and NaCl (1 mL, 1 mmol) EG solutions were subsequently added into PVP solution under strong stirring to form a solution. KF (6 mmol) was also dissolved in EG and added dropwise into the above solution. The mixture was stirred for 1 h, transferred to a polytetrafluoroethylene autoclave, and then heated at 180 °C for 0.5 h. After cooling to room temperature, the products were obtained by centrifugation and washed with deionized water and ethanol several times in order to remove the residual Gd3+ in the solution. The resulting products were dispersed in 10 mL EG as core for further synthesis.
Synthesis of Heterogeneous NaGdF4/ NaYF4:Yb,Er Core/Shell Nanoparticles
PVP (0.5 g) was dissolved in the α-NaGdF4 core (0.5 mmol) solution. Then, YCl3 (0.78 mmol), YbCl3 (0.2 mmol), ErCl3 (0.02 mmol), and NaCl (5 mmol) EG solutions were added respectively under stirring. KF (6 mmol) was dissolved in EG (7 mL) and was subsequently added dropwise into above mixture. After stirring for 1 h, the solution was then transferred into a polytetrafluoroethylene autoclave and reacted at 180 °C for 2 (6, 12, 24) hours. The final product was obtained by centrifugation and washed with ethanol for several times. Half of the final product was dried in vacuum oven at 80 °C for XRD, TEM detection. The other half counterpart was redispersed in water to get the clear solution.
Characterization
X-ray powder diffraction (XRD) analysis was carried out with a powder diffractometer (Model Rigaku RU-200b), using Ni-filtered Cu Kα radiation (λ = 1.5406 Å) with 200 mA current and 50 kV voltage across the tube to generate powerful X-ray. The XRD measurement was performed at a scan rate of 18°min−1 and step size of 0.02°. Scanning electron microscopy (SEM) measurements were carried out by a JEOL JEM-7500F Field Emission SEM. Transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) were recorded on an FEI Tenai F-20 microscopy with a field emission gun operating at 200 kV. Images were acquired digitally by a CCD camera. The local elemental mapping and elemental compositions were determined by energy-dispersive X-ray spectrometry (EDX) under the HR-TEM mode. The UC emission spectra were recorded by a fluorescence spectrometer (Hitachi F-4500) equipped with a 980-nm diode laser. The temporal properties were studied by using a 953.6-nm Raman shifter laser and an oscillograph. The UC luminescent photos of the aqueous solution were taken by a Nikon digital camera (D300s). The digital photographs were taken using identical camera settings and same pumping power. All the measurements were performed at room temperature.
Results and Discussion
In this work, we selected cubic NaGdF4 nanocrystals as cores to induce the growth of hexagonal NaYF4 shells using the heterogeneous core/shell strategy. The heterogeneous-core-mediated method provided a convenient route for facile synthesis of water soluble small-sized hexagonal NaYF4 nanocrystals. Commonly, the NaREF4 crystal exists two polymorphs, cubic and hexagonal phase. The structure of cubic phase NaREF4 is isomorphic with CaF2 (fluorite structure), which contains one type of high-symmetry cation site. In contrast, the crystal structure of hexagonal phase NaREF4 consists of two types of relatively low-symmetry cation sites. In the growth process of NaREF4 crystals, the cubic phase is dynamically stable and preferentially formed. In addition, with a decrease in the particle size, the crystal lattice tends to transform into a structure with higher symmetry. The above nature implies small-sized NaREF4 nanocrystals are cubic phase in general terms and need extra driven force to surpass the energy barrier to realize the transformation from cubic to hexagonal phase. We adopted the cubic heterogeneous core to induce the growth of hexagonal phase shell. The core and shell was different NaREF4. The ionic radii of two kinds of RE ions are closed but different, which means the interfacial energy is small and the heterogeneous core/shell structure can easily be built, but the mismatch intrinsically exists if the heterogeneous interface forms. Due to cation exchange, the heterogeneous interface should be formed when the shell epitaxially grows onto the core [43, 44]. The lattice symmetry of the heterogeneous interface is low because of the mismatch between the core and shell materials, thus triggering the shell subsequently grown with a low symmetric structure, i.e., hexagonal phase. According to the Frank–van der Merwe mode for core–shell heterostructure, once the shell material nucleate heterogeneously onto a pre-existing condensed core, the energy barrier which has to be surpassed is lower than the activation energy which is required to induce the corresponding homogenous nucleation of separate crystal nuclei. It indicates that building a core–shell heterostructure can efficiently decrease the energy barrier, which benefits to the growth of thermodynamically stable β-NaYF4 shell with lower free energy rather than the formation of homogenous α-NaYF4 embryos with higher activation energy. Considering the above factors, we adopt the core/shell heterostructure strategy to control the dynamical process of crystal growth as well as decrease the lattice symmetry of crystal interface to synthesize water soluble β-NaYF4 nanocrystals with small size at low temperature. The schematic diagram of growth process of α-NaGdF4/β-NaYF4:Yb,Er nanocrystals is shown in Fig. 1. In the first step, we synthesized small-sized NaGdF4 nanocrystals by solvothermal reaction at low temperature of 180 °C. Then, we introduced the cubic NaGdF4 nanocrystals into a NaYF4 precursor solution containing Yb3+ and Er3+. In the second solvothermal reaction process, cation exchange firstly occurred. Y3+ entered into the NaGdF4 matrix and exchange with Gd3+ that located in the crystals surface, forming a hetero interface containing both Gd3+ and Y3+ on the surface of the core crystals. Because of the ion radii of Gd3+ and Y3+ are different, it would cause the lattice distortion of the hetero interface, forming a low symmetric structure. Additionally, according to above presentation, the energy barrier of the shell crystals formation could be efficiently decreased by building a heterostructure. Therefore, it would cause the preferential growth of anisotropic hexagonal phase NaYF4 shell with relatively low symmetry following the hetero interface layer by surpassing a lower energy barrier, forming the heterogeneous α-NaGdF4/β-NaYF4:Yb3+,Er3+ core/shell nanoparticles.

Schematic diagram of growth process of heterogeneous core/shell nanocrystals
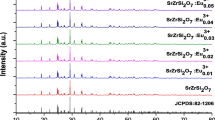
To carry out our idea, we synthesized NaGdF4 core nanocrystals, acting as induced reagents, for the subsequent growth of β-NaYF4 shells. In order to study the growth process of the hexagonal shell, we prepared a set of heterogeneous core/shell crystals by varying the second-step reaction time (shell growth time) from 2 to 24 h. All the reactions in our experiment were based on a solvothermal method using polyvinylpyrrolidone (PVP) as the chelating agent. Both the core and the final core/shell products were examined by XRD to determine the crystal structures. Figure 2 showed the XRD patterns of NaGdF4 core nanocrystals (Fig. 2a) and a set of heterogeneous NaGdF4/NaYF4:Yb,Er core/shell nanocrystals with reaction time of 2, 6, 12, and 24 h (Fig. 2b–e). It can be seen that the core crystals were indexed as pure cubic phase NaGdF4 crystals (Fig. 2g; α-NaGdF4: JCPDS file number 27-697). After subsequent solvothermal treatment with NaYF4 precursors for 2 h, in the XRD pattern (Fig. 2b), there were extra diffraction peaks appeared besides the diffraction peaks of α-NaGdF4. All these new peaks were well consistent with the standard β-NaYF4 crystals (Fig. 2f; β-NaYF4: JCPDS file number 16-334), instead of β–NaGdF4. With the reaction time increasing to 6, 12, and 24 h, the intensities of hexagonal NaYF4 diffraction peaks gradually increased. It indicated that the fraction of the hexagonal phase counterparts in the shells were gradually increased as the reaction proceeding. Note that, for 24-h-reaction samples, the hexagonal NaYF4 diffraction peaks were so intensive that the cubic NaGdF4 peaks were almost not observed (Fig. 2e). The reason may be that the hexagonal NaYF4 shell grew so thick that the strong hexagonal signals pressed down the cubic phase signals, leading to the seemed-absence of α-NaGdF4 diffraction peaks. In a word, the XRD results indicated the formation of hexagonal NaYF4 crystals which become more and more dominant with the reaction time increasing.

XRD patterns of (a) the NaGdF4 core nanocrystals, and the NaGdF4/ NaYF4:Yb,Er core/shell (named C/S in the figure) nanocrystals with different reaction time, (b) 2 h, (c) 6 h, (d) 12 h, and (e) 24 h. (f) Standard data of β-NaYF4 (JCPDS 16-334). (g) Standard data of α-NaGdF4 (JCPDS 27-697)
We also performed another set of contrast experiments that we synthesized the NaYF4 crystals without α-NaGdF4 core existing, keeping all other reaction conditions the same as the preparation of NaGdF4/NaYF4 nanocrystals. The crystal structures were examined by XRD. As shown in Fig. 3a, the diffraction peaks of the 2-h-reaction NaYF4:Yb,Er crystals were indexed to cubic NaYF4 crystals (JCPDS file number 77-2042). With reaction time increasing to 6 and 12 h, even to 24 h (Fig. 3b–d), the as-prepared NaYF4 crystals still keep the pure cubic phase, instead of appearance of any hexagonal phase NaYF4 signals. These results indicated that, in this low-temperature solvothermal reaction system, the α–NaGdF4 core crystals were the requirement for growth of hexagonal phase NaYF4 crystals.

XRD patterns of NaYF4: Yb, Er crystals which were synthesized without α–NaGdF4 core existing with different reaction times. (a) 2 h, (b) 6 h, (c) 12 h, and (d) 24 h
The morphologies and size distributions of the core and core/shell samples are shown in Fig. 4. We performed TEM to characterize the morphologies of the cubic NaGdF4 core nanocrystals (Fig. 4A (a)) and heterogeneous NaGdF4/NaYF4 core/shell crystals with the shell growth time of 2, 6, 12, and 24 h, respectively (Fig. 4A (b–f)). The corresponding size distributions were shown in Fig. 4B. From the TEM image of the cubic NaGdF4 core nanocrystals (Fig. 4A (a)), we can see that the particles were nearly uniform square shape, with average size about 23 nm (corner to corner), which was shown in the histogram of size distribution (Fig. 4B (m)). The HRTEM image displayed the clear lattice fringes pattern of the core crystals (Fig. 4A (h)), and the interplanar spacing of 0.31 nm was indexed to (111) plane of cubic NaGdF4. After subsequent growth of NaYF4 shell for 2 h, the particle shapes varied that the nanosquare edges became somewhat rounded and the particle size increased to 28 nm on average (Fig. 4B (n)). The lattice fringes were shown in HRTEM image, in which the measured interplanar spacing of 0.299 nm was indexed to (110) plane of hexagonal NaYF4 crystal. Combining the TEM, HRTEM, and XRD results, for 2-h-reaction samples, the growth of NaYF4 shell with thickness of 2.5 nm and the appearance of hexagonal NaYF4 lattice fringes and diffraction peaks could preliminarily confirmed the formation of hexagonal NaYF4 shells on the cubic NaGdF4 cores. Further prolonging the reaction time to 6 and 12 h, the particle size continued increasing to 33 and 38 nm, respectively, and the particle shape varied to nanopolyhedron, enclosed by the (101) and (100) planes, respectively (Fig. 4A (j, k)). Note that the increase of shell thickness were in agreement with the data in XRD that the hexagonal NaYF4 diffraction peaks became more and more intensive, which further implied that the NaYF4 crystals existed in the form of hexagonal shell. Then, extending the reaction time to 24 h, we found that, from 12 to 24 h, the crystals grew rapidly to the hexagonal prism shape, which is shown in the TEM images (Fig. 4A (e, f)) and SEM image (Fig. 4A (g)). We can see the hexagonal nanoprism stood on the TEM grids either on their bottom faces or on their side faces. The mean size was about 115 nm × 125 nm (diameter from corner to corner × height, shown in Fig. 4B (q, r)) and the hexagonal NaYF4 lattice fringes were also displayed clearly in HRTEM image (Fig. 4A (l)) that the interplanar spacing of 0.519 nm was indexed to (100) plane. For the 24-h-reaction heterogeneous NaGdF4/NaYF4:Yb,Er core/shell samples, the shell grew so thick (~ 50 nm) that, consistently, in above XRD pattern (Fig. 2e), the hexagonal NaYF4 peaks were dominant and the cubic NaGdF4 peaks almost cannot be observed.

A TEM and HRTEM images of the as-prepared cubic core crystals and heterogeneous core/shell crystals. TEM images of (a) cubic NaGdF4 core nanocrystals, (b)–(e) heterogeneous NaGdF4/ NaYF4 core/shell crystals with the shell growth time of 2, 6, 12, and 24 h, respectively, (f) a single particle of the 24-h-reaction NaGdF4/ NaYF4 core/shell crystals which stand on the TEM grids on their bottom faces. (g) SEM image of the 24-h-reaction NaGdF4/NaYF4 core/shell crystals, which exhibit a hexagonal nanoprism look. HRTEM images of (h) cubic NaGdF4 core nanocrystals. (i)–(l) heterogeneous NaGdF4/NaYF4 core/shell crystals with the shell growth time of 2, 6, 12, and 24 h, respectively. B Histograms of size distribution of (m) cubic NaGdF4 core nanocrystals, (n)–(p) heterogeneous NaGdF4/NaYF4 core/shell crystals with the shell growth time of 2, 6, and 12 h, respectively, (q) and (r) diameter of the hexagonal cross section and the nanoprism height of the 24-h-reaction NaGdF4/ NaYF4 core/shell crystals
EDX analysis was performed to examine the elemental composition of the heterogeneous NaGdF4/NaYF4 core/shell crystals. Figure 5 showed the EDX pattern of 6-h-reaction samples. The major elements of Gd, Y, Yb, Na, and F were observed in the pattern, while Er element was not observed because of the ultra-little dopant concentration of 2%.

EDX analysis of elemental composition of heterogeneous NaGdF4/ NaYF4 core/shell crystals
To further confirm the heterogeneous core/shell geometry of the as-prepared α-NaGdF4/β-NaYF4 crystals, the local elemental mapping by EDX line scans were carried out to address the composition across of the nanocrystals. The images of EDX elemental mapping are given in Fig. 6, in which (a)–(d) were the image of selected scan area, Gd, Y, and Yb, respectively, of the 2-h-reaction samples, and (e)–(h) were the image of selected scan area, Gd, Y, and Yb, respectively, of the 6-h-reaction samples. For the two crystal samples, we observed that all the three examined elements of Gd, Y, and Yb are homogenously dispersed overlap the nanocrystals, and the labeled areas of Gd element were a little smaller than that of Y and Yb elements. The fact suggested that Y and Yb elements uniformly covered across the whole outer layer of the nanocrystals, while Gd element was localized in the center. These elemental mapping results were strong proofs of the heterogeneous core/shell structure formation of the α-NaGdF4/β-NaYF4:Yb,Er crystals.

Images of local elemental mapping by EDX line scans for a–d the 2-h-reaction NaGdF4/NaYF4:Yb,Er crystals and e–h the 6-h-reaction NaGdF4/NaYF4:Yb,Er crystals. a, e image of EDX line scans areas. b, f Gd in core. c, g Y in shell. d, h Yb in shell
Next, the optical properties of the heterogeneous α-NaGdF4/ β-NaYF4: Yb,Er core/shell crystals were characterized and discussed. Co-doping Yb3+ (20 mol%) and Er3+ (2 mol%) in the hexagonal NaYF4 shell could realize efficient UC luminescence. Under 980-nm excitation, Yb3+ ions can absorb infrared photons and transfer the energy to Er3+ ions successively. The excited Er3+ ions then emit characteristic UC luminescence in the region of 300–700 nm. Figure 7 showed the UC luminescence spectra of the x-hours-reaction (x =changed the NaREF4 to other 2, 6, 12, 24) α-NaGdF4/β-NaYF4: Yb, Er crystals under 980-nm excitation, with the same pumping power of 160 mW (pumping power density of 16 W cm−2). In each spectrum of the as-prepared samples, there were all the characteristic UC peaks of Er3+ ions. We observed that with the shell growth time increasing, the overall emission intensities increased, while the intensity ratios of every emission peaks almost unchanged. The green emission peaks in the range of 515–560 nm from the 2H11/2, 4S3/2 → 4I15/2 transitions were relatively strong, while the blue emission peaks from the 2H9/2 → 4I15/2 transitions centered at 409 nm and the red emission peaks between 640 and 680 nm, corresponding to the 4F9/2 → 4I15/2 transitions, were relatively weak. Interestingly, several so-weak emission peaks centered at 317, 312, and 277 nm were observed, which originated from Er3+: 4P3/2 → 4I15/2, Gd3+: 6P7/2 → 8 s7/2, and Gd3+ [6]:IJ → 8 s7/2 (inset of Fig. 7). It is known that Gd3+ ions cannot absorb 980-nm photons directly; therefore, there must exist energy transfer processes from Er3+ to Gd3+ at high energy excited states, which induced the UC emissions of Gd3+ ions. In our designed core/shell α-NaGdF4/β-NaYF4: Yb, Er crystals, Gd3+ ions were located in the core and Yb3+, Er3+ ions existed in the shell; thus reasonably, the energy transfer processes from Er3+ to Gd3+ should occur in the hetero interface between the α-NaGdF4 core and β-NaYF4 shell. Inspiringly, this is a proof to certify the existence of the hetero interface which attributed to the hexagonal shell growth in our reasoning.

UC luminescence spectra of the as-prepared heterogeneous core/shell α-NaGdF4/β-NaYF4:Yb,Er crystals with varied reaction time (black: 2 h, red: 6 h, green: 12 h, blue: 24 h) excited by 980-nm infrared light. (Inset: Local magnified patterns of UC spectra in the region of 265–330 nm, in which the characteristic peaks of Gd3+ were observed)
In addition, we measured the lifetimes of various levels of Yb3+ and Er3+ in x-hours-reaction (x = 2, 6, 12, 24) α-NaGdF4/β-NaYF4:Yb,Er core/shell crystals, using a 953.6-nm pulsed laser with a pulse width of 10 ns and a repetition rate of 10 Hz as the excitation source. Figure 8 showed the photoluminescence decay curves of the 2F5/2 level of Yb3+ (Fig. 8a) and 4F9/2, 4F9/2, 2H9/2 levels of Er3+ (Fig. 8b–d) in the x-hours-reaction (x = 2, 6, 12, 24) α-NaGdF4/β-NaYF4: Yb, Er core/shell crystals. All the decay curves could be well fitted by a single exponential function I (t) = I 0 exp.(−t/τ), where I 0 is the initial emission intensity at t = 0 and τ is the lifetime of the monitored level. The measured lifetime data of all monitored levels in each detected samples were listed in Fig. 8. Obviously, for all monitored levels, the lifetime gradually increased with the shell growth time increasing from 2, to 6, to 12, to 24 h. For example, the measured lifetime of 4F9/2 level of Er3+ is 54, 109, 139, and 259 μs for 2, 6, 12, and 24-h-reaction α-NaGdF4/β-NaYF4: Yb, Er core/shell crystals, respectively (Fig. 8c). These results were consistent with the UC luminescence spectra data that the overall emission intensities increased with the shell growth time increasing. The reason was that the β-NaYF4: Yb, Er shell counterpart which provided UC luminescence was increased with the reaction being prolonged.

Photoluminescence decay curves of the a 2F5/2 level of Yb3+, b 4F9/2 level of Er3+, c 4F9/2 level of Er3+, and d 2H9/2 level of Er3+ in the heterogeneous core/shell α-NaGdF4/β-NaYF4:Yb, Er crystals with varied reaction time (black: 2 h, red: 6 h, blue: 12 h, pink: 24 h)
Conclusions
In summary, a heterogeneous core/shell structure was constructed, in which cubic NaGdF4 nanocrystals, serving as cores, induced the growth of the heterogeneous hexagonal NaYF4 shells co-doped with Yb3+ and Er3+ ions. We prepared a set of heterogeneous α-NaGdF4/β-NaYF4:Yb,Er core/shell particles by varying the shell growth time from 2 to 24 h. The characterizations of XRD, TEM, HRTEM, EDX, and local elemental mapping results were performed. With the growth time increasing from 2 h, to 6 h, to 12 h, and finally to 24 h, the diffraction peaks of hexagonal NaYF4 in the XRD patterns became more and more dominant, and the crystals shapes changed from nanosquare, to nanopolyhedron, and finally to hexagonal prism shape, and the particle size increased from 28 nm, to 33 nm, to 38 nm, and finally to 115 nm × 125 nm, respectively. Moreover, in the local elemental mapping images, Gd element was localized in the center, and Y and Yb elements uniformly covered across the whole outer layer of the nanocrystals. All the results confirmed the successful formation of the unique heterogeneous α-NaGdF4/β-NaYF4:Yb,Er core/shell particles. Moreover, the existence of hetero interface between cubic cores and hexagonal shells was confirmed by the observation of Gd3+ UC emission. In our reasoning, we demonstrated that the hetero interface, which were produced by cation exchanges that caused a lower lattice symmetric structure, attributed to the heterogeneous hexagonal shell growth.
References
Auzel F (2004) Upconversion and anti-stokes processes with f and d ions in solids. Chem Rev 104:139–174
Wang L, Yan R, Huo Z, Wang L, Zeng J, Bao J, Wang X, Peng Q, Li Y (2005) Fluorescence resonant energy transfer biosensor based on upconversion-luminescent nanoparticles. Angew Chem Int Ed 44:6054–6057
Heer S, Kömpe K, Güdel HU, Haase M (2004) Highly efficient multicolour upconversion emission in transparent colloids of lanthanide-doped NaYF4 nanocrystals. Adv Mater 16:2102–2105
Scheps R (1996) Upconversion laser processes. Prog Quantum Electron 20:271–358
Pollnau M, Gamelin DR, Lüthi SR, Güdel HU, Hehlen MP (2000) Power dependence of upconversion luminescence in lanthanide and transition-metal-ion systems. Phys Rev B 61:3337–3346
Li Z, Zhang Y, Jiang S (2008) Multicolor core/shell-structured upconversion fluorescent nanoparticles. Adv Mater 20:4765–4769
Heer S, Lehmann O, Haase M, Güdel H-U (2003) Blue, green, and red upconversion emission from lanthanide-doped LuPO4 and YbPO4 nanocrystals in a transparent colloidal solution. Angew Chem Int Ed 42:3179–3182
Wang X, Liu Q, Bu Y, Liu C-S, Liu T, Yan X (2015) Optical temperature sensing of rare-earth ion doped phosphors. RSC Adv 5:86219–86236
Xu J, Zhou S, Tu D, Zheng W, Huang P, Li R, Chen Z, Huang M, Chen X (2016) Sub-5 nm lanthanide-doped lutetium oxyfluoride nanoprobes for ultrasensitive detection of prostate specific antigen. Chem Sci 7:2572–2578
Chatterjee DK, Rufaihah AJ, Zhang Y (2008) Upconversion fluorescence imaging of cells and small animals using lanthanide doped nanocrystals. Biomaterials 29:937–943
Zhou J, Liu Z, Li F (2012) Upconversion nanophosphors for small-animal imaging. Chem Soc Rev 41:1323–1349
Idris NM, Gnanasammandhan MK, Zhang J, Ho PC, Mahendran R, Zhang Y (2012) In vivo photodynamic therapy using upconversion nanoparticles as remote-controlled nanotransducers. Nat Med 18:1580–1585
Zhou J, Sun Y, Du X, Xiong L, Hu H, Li F (2010) Dual-modality in vivo imaging using rare-earth nanocrystals with near-infrared to near-infrared (NIR-to-NIR) upconversion luminescence and magnetic resonance properties. Biomaterials 31:3287–3295
Cheng L, Yang K, Li Y, Chen J, Wang C, Shao M, Lee S-T, Liu Z (2011) Facile preparation of multifunctional upconversion nanoprobes for multimodal imaging and dual-targeted photothermal therapy. Angew Chem 123:7523–7528
Wang F, Liu X (2009) Recent advances in the chemistry of lanthanide-doped upconversion nanocrystals. Chem Soc Rev 38:976–989
Zeng JH, Su J, Li ZH, Yan RX, Li YD (2005) Synthesis and upconversion luminescence of hexagonal-phase NaYF4:Yb, Er3+ phosphors of controlled size and morphology. Adv Mater 17:2119–2123
Suyver JF, Grimm J, van Veen MK, Biner D, Krämer KW, Güdel HU (2006) Upconversion spectroscopy and properties of NaYF4 doped with Er3+, Tm3+ and/or Yb3+. J Lumin 117:1–12
Liu Q, Sun Y, Yang T, Feng W, Li C, Li F (2011) Sub-10 nm hexagonal lanthanide-doped NaLuF4 upconversion nanocrystals for sensitive bioimaging in vivo. J Am Chem Soc 133:17122–17125
Zhai X, Liu S, Zhang Y, Qin G, Qin W (2014) Controlled synthesis of ultrasmall hexagonal NaTm0.02Lu0.98−xYbxF4 nanocrystals with enhanced upconversion luminescence. J Mater Chem C 2:2037
Mi C, Tian Z, Cao C, Wang Z, Mao C, Xu S (2011) Novel microwave-assisted solvothermal synthesis of NaYF4:Yb,Er upconversion nanoparticles and their application in cancer cell imaging. Langmuir 27:14632–14637
Yi G-S, Chow G-M (2007) Water-soluble NaYF4:Yb,Er(tm)/NaYF4/polymer core/shell/shell nanoparticles with significant enhancement of upconversion fluorescence. Chem Mater 19:341–343
Hassan SMu, Kitamoto Y (2015) Facile fabrication of porous hollow upconversion capsules using hydrothermal treatment. Mater Chem Phys 167:49–55
Zhuang L, Sung HHY, Yang W, Williams ID, Feng QS (2007) Controlled hydrothermal growth and up-conversion emission of NaLnF4 (Ln = Y, Dy−Yb). Inorg Chem 46:5404–5410
Wang F, Liu X (2008) Upconversion multicolor fine-tuning: visible to near-infrared emission from lanthanide-doped NaYF4 nanoparticles. J Am Chem Soc 130:5642–5643
Boyer J-C, Vetrone F, Cuccia LA, Capobianco JA (2006) Synthesis of colloidal upconverting NaYF4 nanocrystals doped with Er3+, Yb3+ and Tm3+, Yb3+ via thermal decomposition of lanthanide trifluoroacetate precursors. J Am Chem Soc 128:7444–7445
Haase M, Schafer H (2011) Upconverting nanoparticles. Angew Chem Int Ed Engl 50:5808–5829
Mai H-X, Zhang Y-W, Si R, Yan Z-G, Sun L-d, You L-P, Yan C-H (2006) High-quality sodium rare-earth fluoride nanocrystals: controlled synthesis and optical properties. J Am Chem Soc 128:6426–6436
Xue X, Wang L, Huang L, Zhao D, Qin W (2013) Effect of alkali ions on the formation of rare earth fluoride by hydrothermal synthesis: structure tuning and size controlling. CrystEngComm 15:2897
Chen D, Yu Y, Huang F, Huang P, Yang A, Wang Y (2010) Modifying the size and shape of monodisperse bifunctional alkaline-earth fluoride nanocrystals through lanthanide doping. J Am Chem Soc 132:9976–9978
Wang X, Bu Y, Xiao Y, Kan C, Lu D, Yan X (2013) Size and shape modifications, phase transition, and enhanced luminescence of fluoride nanocrystals induced by doping. J Mater Chem C 1:3158–3166
Wang F, Han Y, Lim CS, Lu Y, Wang J, Xu J, Chen H, Zhang C, Hong M, Liu X (2010) Simultaneous phase and size control of upconversion nanocrystals through lanthanide doping. Nature 463:1061–1065
Chen H, Zhai X, Li D, Wang L, Zhao D, Qin W (2012) Water-soluble Yb3+, Tm3+ codoped NaYF4 nanoparticles: synthesis, characteristics and bioimaging. J Alloys Compd 511:70–73
Wang G, Qin W, Wang L, Wei G, Zhu P, Kim R (2008) Intense ultraviolet upconversion luminescence from hexagonal NaYF4:Yb3+/Tm3+ microcrystals. Opt Express 16:11907–11914
Wang Y, Cai R, Liu Z (2011) Controlled synthesis of NaYF4: Yb, Er nanocrystals with upconversion fluorescence via a facile hydrothermal procedure in aqueous solution. Cryst Eng Comm 13:1772–1774
Shi F, Zhao Y (2014) Sub-10 nm and monodisperse β-NaYF4:Yb,tm,Gd nanocrystals with intense ultraviolet upconversion luminescence. J Mater Chem C 2:2198
Liu D, Xu X, Du Y, Qin X, Zhang Y, Ma C, Wen S, Ren W, Goldys EM, Piper JA, Dou S, Liu X, Jin D (2016) Three-dimensional controlled growth of monodisperse sub-50 nm heterogeneous nanocrystals. Nat Commun 7:10254
Boyer JC, Manseau MP, Murray JI, van Veggel FC (2010) Surface modification of upconverting NaYF4 nanoparticles with PEG-phosphate ligands for NIR (800 nm) biolabeling within the biological window. Langmuir 26:1157–1164
Chatterjee DK, Fong LS, Zhang Y (2008) Nanoparticles in photodynamic therapy: an emerging paradigm. Adv Drug Deliv Rev 60:1627–1637
Zhao D, Chen H, Zheng K, Chuai X, Yu F, Li H, Wu C, Qin G, Di W, Qin W (2014) Growth of hexagonal phase sodium rare earth tetrafluorides induced by heterogeneous cubic phase core. RSC Adv 4:13490
Chen Q, Xie X, Huang B, Liang L, Han S, Yi Z, Wang Y, Li Y, Fan D, Huang L, Liu X (2017) Confining excitation energy in Er3+-sensitized upconversion nanocrystals through Tm3+-mediated transient energy trapping. Angew Chem Int Ed 56:7605–7609
Wang X, Liu Q, Cai P, Wang J, Qin L, Vu T, Seo HJ (2016) Excitation powder dependent optical temperature behavior of Er3+ doped transparent Sr0.69La0.31F2.31 glass ceramics. Opt Express 24:17792–17804
Wang X, Bu Y, Yan X, Cai P, Wang J, Qin L, Vu T, Seo HJ (2016) Detecting the origin of luminescence in Er3+-doped hexagonal Na1.5Gd1.5F6 phosphors. Opt Lett 41:5314–5317
Dong C, Korinek A, Blasiak B, Tomanek B, van Veggel FCJM (2012) Cation exchange: a facile method to make NaYF4:Yb,Tm-NaGdF4 core–shell nanoparticles with a thin, tunable, and uniform shell. Chem Mater 24:1297–1305
Mews A, Eychmueller A, Giersig M, Schooss D, Weller H (1994) Preparation, characterization, and photophysics of the quantum dot quantum well system cadmium sulfide/mercury sulfide/cadmium sulfide. J Phys Chem 98:934–941
Acknowledgements
This work was supported by China Postdoctoral Science Foundation (2016M591475).
Author information
Authors and Affiliations
Contributions
HC and DZ provided the ideas and structure of the whole article. PZ mainly completed the “Methods” section. HC and WQ wrote “optical properties” section. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Chen, H., Zhang, P., Cui, H. et al. Synthesis and Luminescence Properties of Water Soluble α-NaGdF4/β-NaYF4:Yb,Er Core–Shell Nanoparticles. Nanoscale Res Lett 12, 548 (2017). https://doi.org/10.1186/s11671-017-2306-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s11671-017-2306-3